
Genetics and Disease — MCQs

On this page
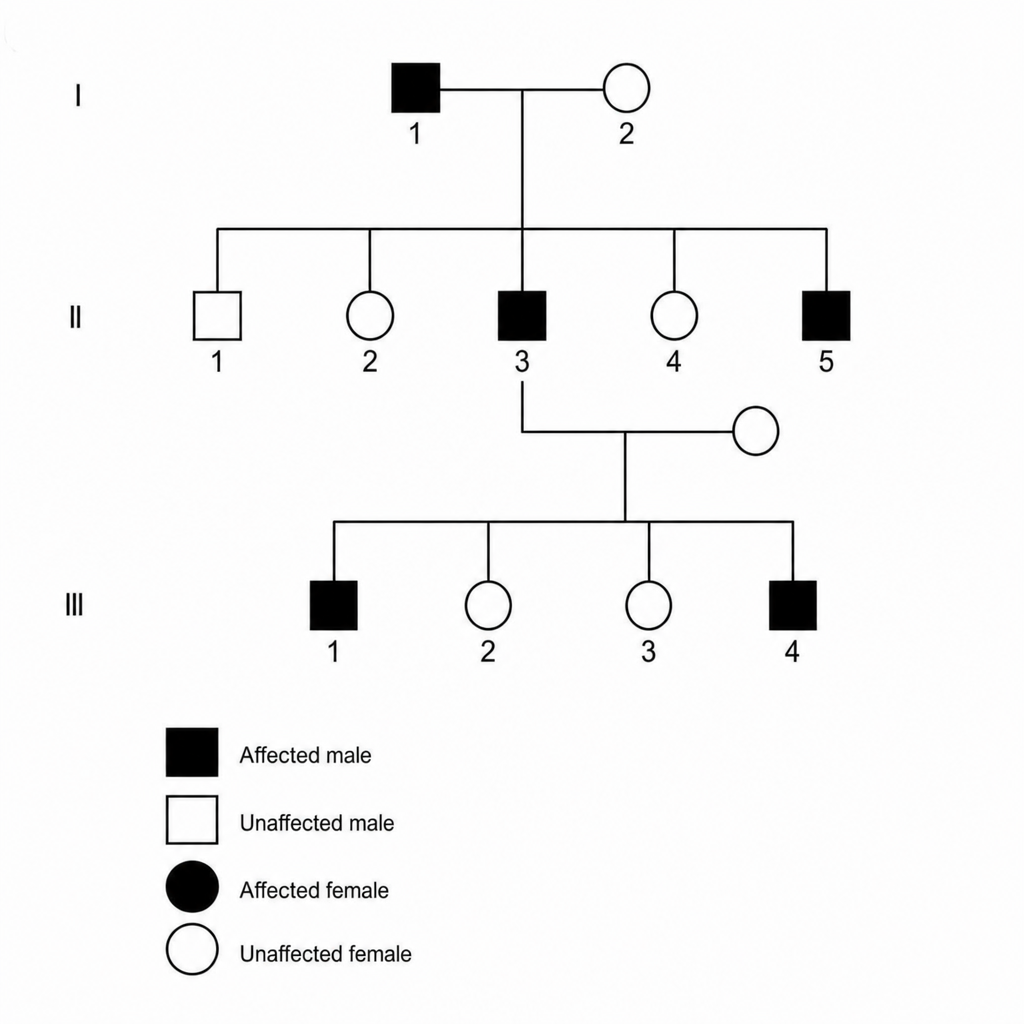
A 25-year-old man presents for a routine physical examination. The patient is tall (6 ft. 5 in) and on examination he was found to have an early diastolic murmur. His family pedigree is as given below. Which of the following is the mode of inheritance by which the disease is likely to be transmitted?
A patient with hemochromatosis experiences joint pain and fatigue. Which genetic test would confirm the diagnosis?
A 35-year-old woman presents with hepatomegaly and arthritis. Laboratory results show high serum ferritin and transferrin saturation. Which genetic defect is likely?
Which genetic syndrome is associated with hundreds of colorectal polyps and almost certain progression to colorectal cancer if left untreated?
A 25-year-old with Gaucher disease type 1 and a GBA mutation, exhibiting significant residual β-glucocerebrosidase activity, what is the best initial treatment?
Which of the following is NOT a feature of Abetalipoproteinemia?
Following genetic counselling in a family for Familial polyposis coli (FPC), what is the next appropriate screening test for at-risk individuals?
Which karyotype is commonly associated with true hermaphroditism?
A 25-year-old man presents for a routine physical examination. He is tall and has an early diastolic murmur on examination. Based on the clinical findings, which of the following is the most likely mode of inheritance for the condition?
Which one of the following is an autosomal recessive disorder?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app