
Genetics and Disease — MCQs

On this page
Which pattern of inheritance of disease is associated with consanguinity?
Which of the following provide protection against malaria except
Syndrome which is characterized by 2X chromosomes and 1Y chromosome is:
Malaria protection comes from all Except
Which of the following is a sex linked inherited disease?
A patient taken for surgery was given suxamethonium, following which his body temperature began to rise quickly. Along with this he also developed masseter spasm, tachycardia and arrhythmia. An ABG analysis also revealed that he has hypoxia, hypercarbia and acidosis. What is the mode of inheritance of the condition described above?
Which one of the following is NOT a contraindication in Porphyria?
A 15 year old child has developed dystonia and poor school grades. Slit examination is shown below. Which is the initial investigation recommended for the patient ?
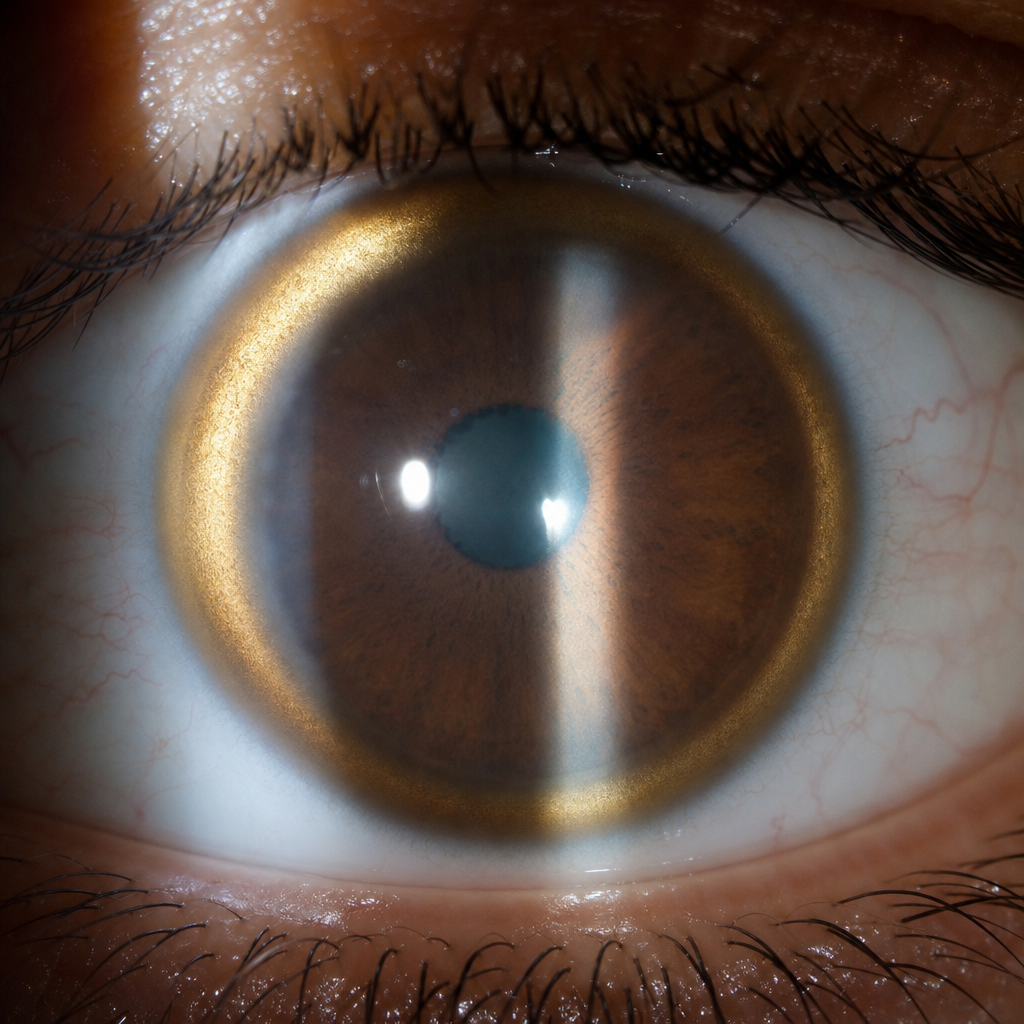
A 60-year-old man presents with choreoathetosis. On examination, Kayser-Fleischer rings are seen in the cornea. What is the initial treatment of choice for this condition?
Which of the following is autosomal dominant in inheritance?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app