
Genetics and Disease — MCQs

On this page
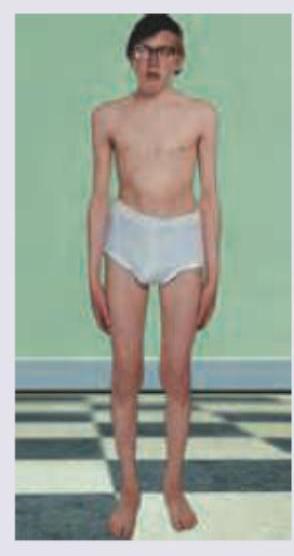
A teenage boy, who was previously raised as a girl, presents with primary amenorrhea and lack of secondary sexual characteristics. On examination, he has tall stature, small testes, and gynecomastia. Which of the following is the most likely genotype?
Which of the following genetic mutations is most commonly associated with familial cases of Amyotrophic Lateral Sclerosis (ALS)?
The child is having skeletal abnormality in arms and hands. Which possible heart disease is seen in this condition?
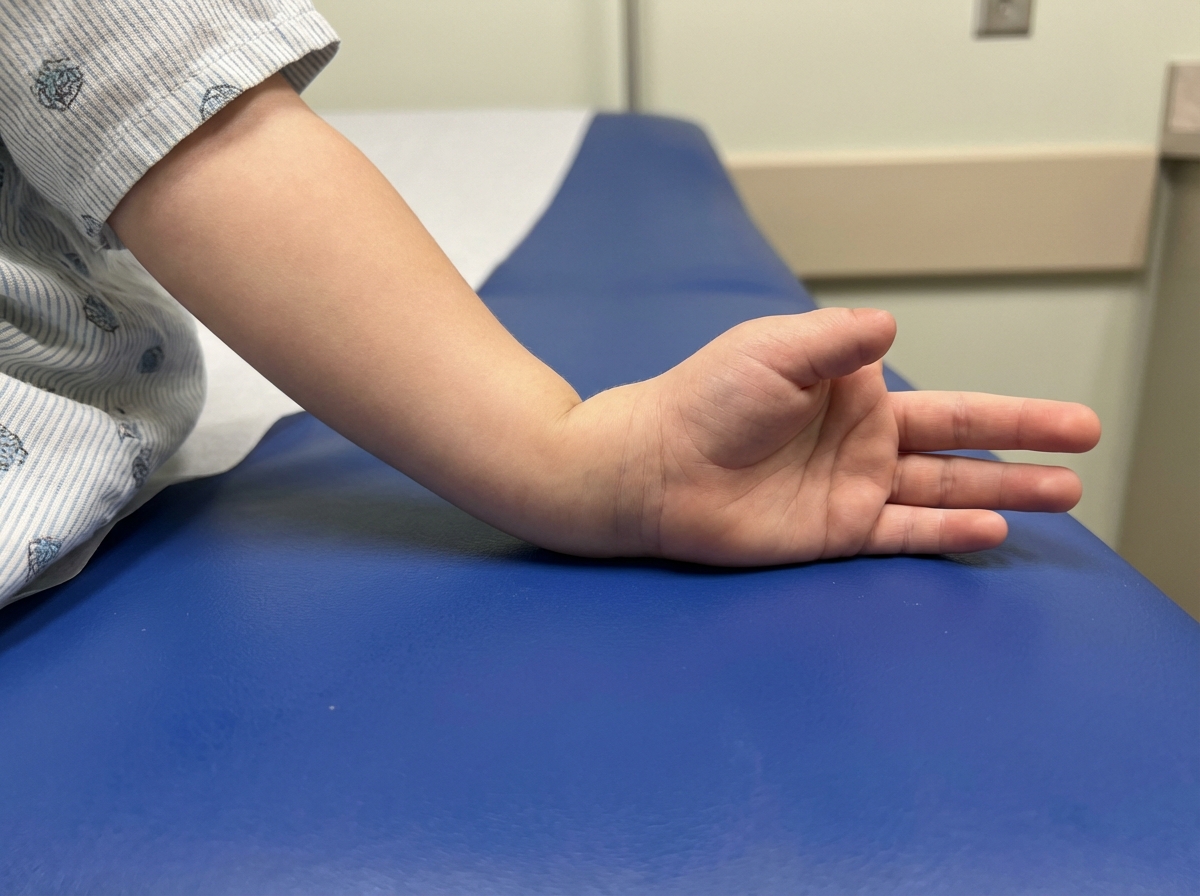
The image reveals hyper-extensible skin seen with Ehlers-Danlos syndrome. Which of the following is commonly seen in Marfan syndrome?
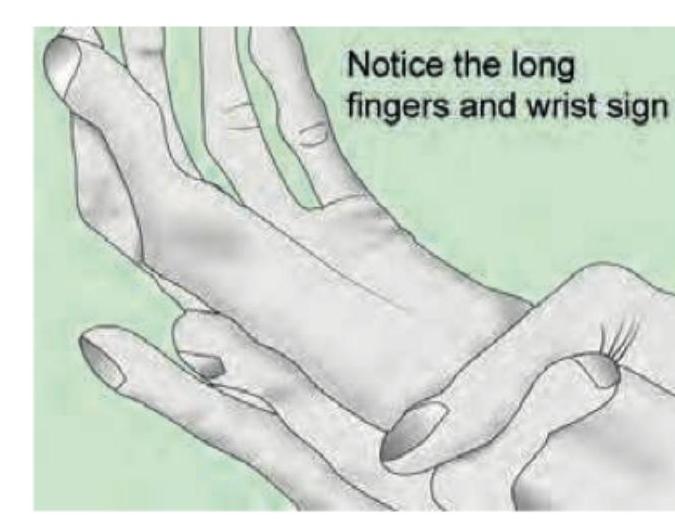
All are correct about this patient except:
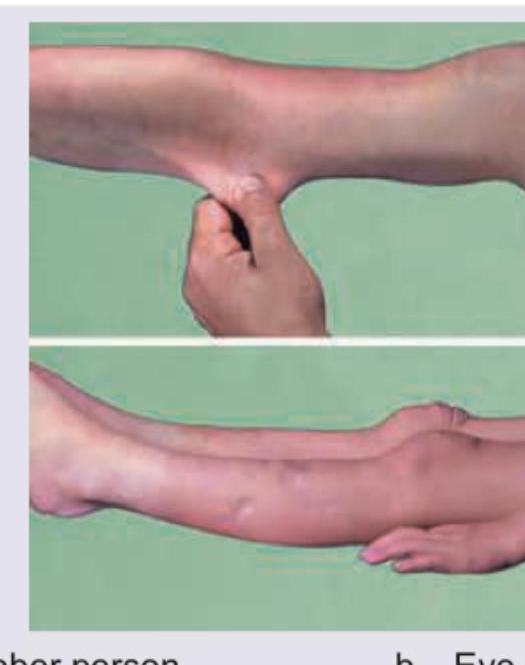
Which of the following will not be the feature of this patient shown below who has urine nitroprusside test positive? (Recent NEET Pattern 2016-17)
A patient presents with headache, confusion, and a diagnosis of a brain tumor. The family history reveals brain and kidney tumors. What is the most likely diagnosis?
Seminal vesicles and vas deferens would be bilaterally absent congenitally in which of the following conditions?
Which of the following statements are true about familial adenomatous polyposis? 1. It is autosomal recessive 2. If not treated, 100% of the cases progress to adenocarcinoma colon. 3. It is associated with a gene mutation in KRAS 4. It is associated with congenital hypertrophy of the retinal pigment epithelium.
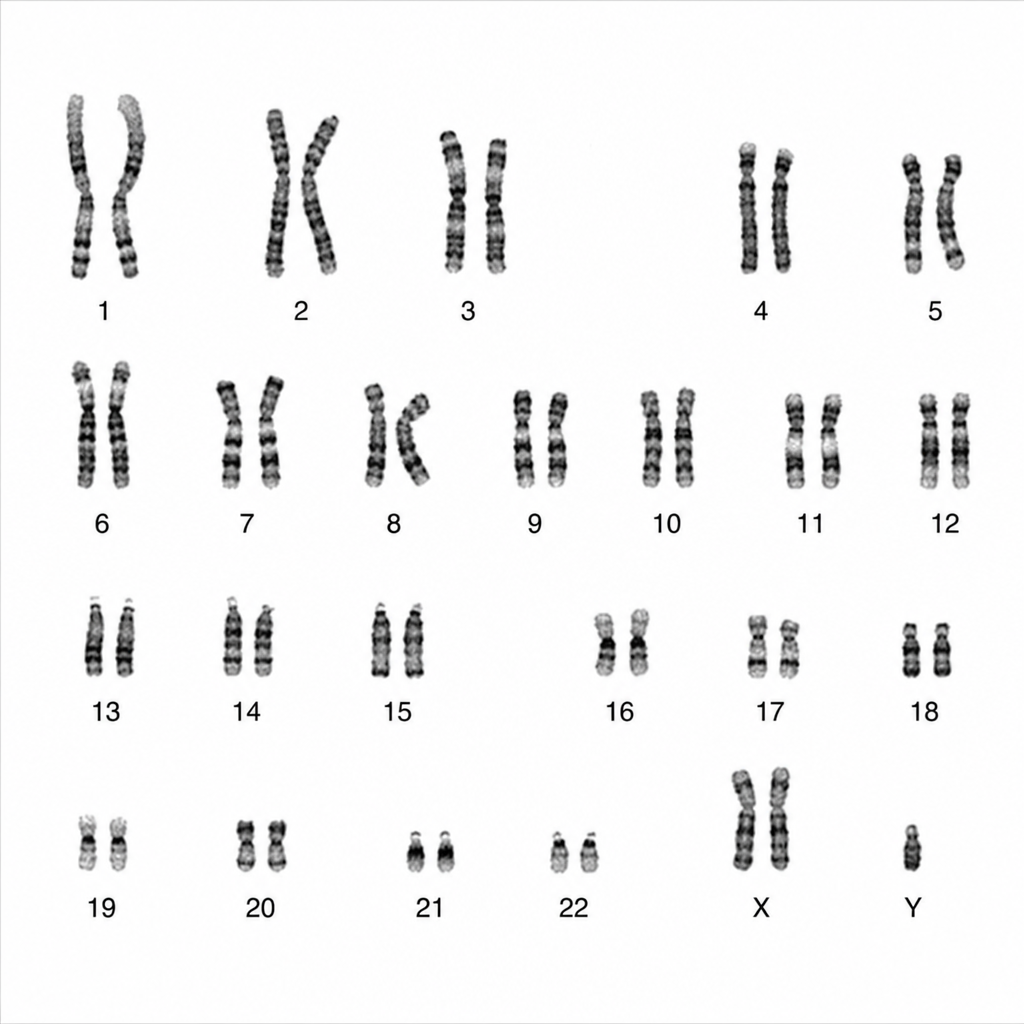
A karyotyping image is given below. What are the clinical features most likely to be expected in such patients?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app