
Genetics and Disease — MCQs

On this page
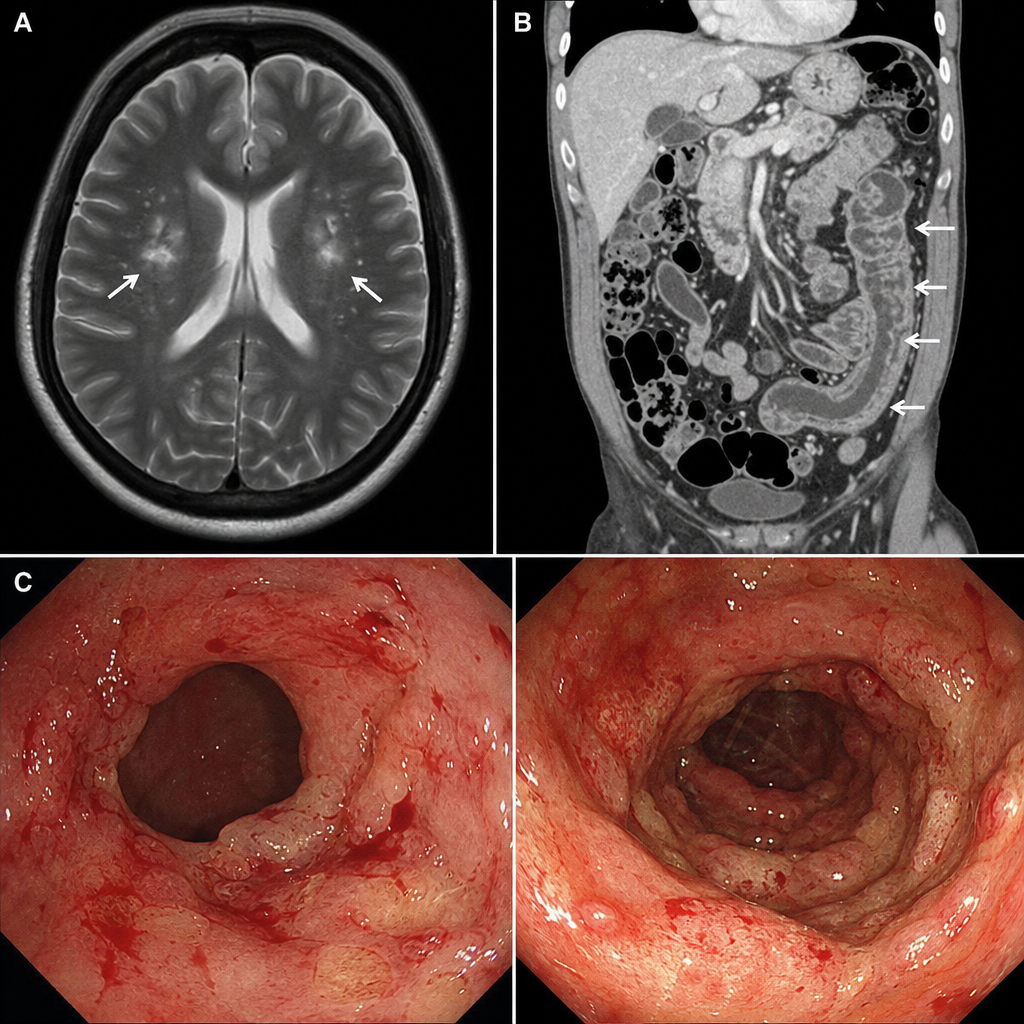
A 40-year-old male presented to the ER with generalized tonic-clonic seizure. This was his first episode and he gave a history of intermittent bloody stools for the past 5 months. Investigations included MRI scan of the head, CECT abdomen, and colonoscopy. What is the most common inheritance pattern of the condition suggested by this presentation?
Which of the following is NOT a mitochondrial disease?
Which of the following conditions is inherited in an autosomal dominant pattern?
A 32-year-old woman presents with a 2-day history of decreased vision and photopsia in her left eye. She is noted to be unusually tall with long limbs and slender fingers. Physical examination reveals pectus excavatum, kyphoscoliosis, a midsystolic click, and a pansystolic murmur. Ophthalmologic examination shows a retinal detachment and lens dislocation. The patient is at greatest risk for which of the following conditions?
A very tall, slender 16-year-old boy is referred for evaluation of an abnormal CXR. He reports no pulmonary or cardiac symptoms and feels well. On physical examination, he has long fingers, pectus excavatum, and a high arched palate. Which of the following is most likely to be seen on his CXR?
A 32-year-old man presents for routine evaluation. He has no symptoms but has noticed some new "nodules" on his legs. Physical examination reveals lumps on his Achilles tendon, yellow lesions around his eyes, and pigmentation of his iris. Which of the following is the most likely diagnosis?
A 20-year-old female on oral contraceptive pills presents with behavioral changes and abdominal pain. What is the most probable diagnosis?
A female child presents with virilization and hypertension with low plasma renin. What is the most likely diagnosis?
Which of the following is NOT a characteristic of Xeroderma Pigmentosum?
In a family, the father has widely spaced eyes, increased facial hair, and deafness. One of the three children has deafness with similar facial features. The mother is normal. Which one of the following is the most likely pattern of inheritance in this case?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app