
Genetics and Disease — MCQs

On this page
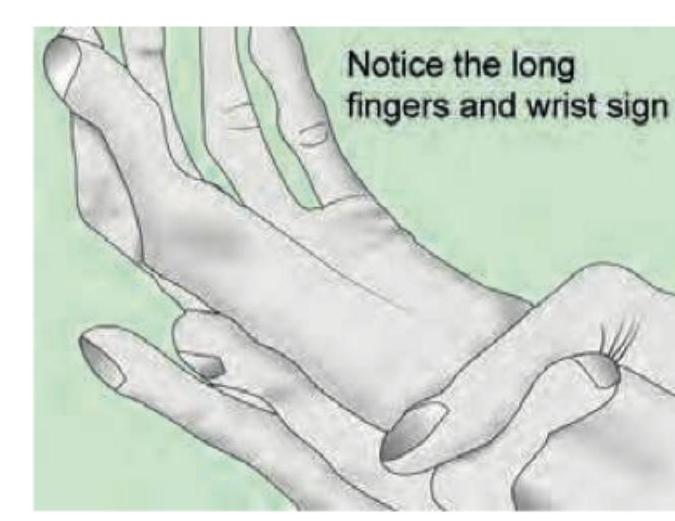
A patient has a long arm span, hypermobile joints and ectopia lentis. What is the defective protein?
Genetic testing for BRCA 1/BRCA 2 is indicated for all of the following except:
Which gene would you test if the patient has a family history of breast and ovarian cancer?
The pedigree diagram of a family is shown below. Affected individuals present with progressive external ophthalmoplegia, pigmentary retinopathy, and cardiac conduction defects. Based on the pedigree and clinical features, what is the most likely diagnosis?
A patient presents with orange-colored tonsils. Laboratory investigations reveal triglyceride level of 140 mg/dL and HDL cholesterol of 5 mg/dL. What is the most likely diagnosis?
A patient has a family history positive for premature coronary artery disease. All are true about the disease shown in the image, except?
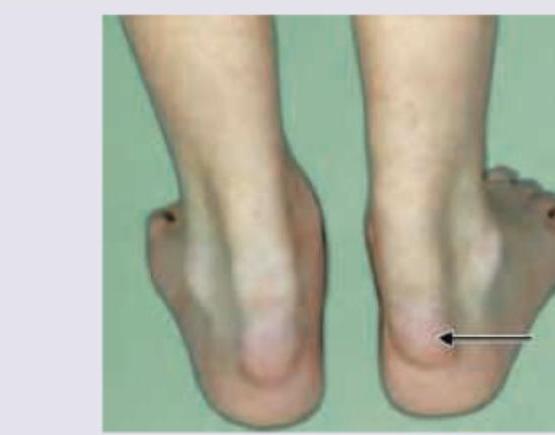
A 32-year-old woman with a history of multiple hamartomas scattered throughout the small intestine presents to a physician for follow-up. All are true about her diagnosis except?
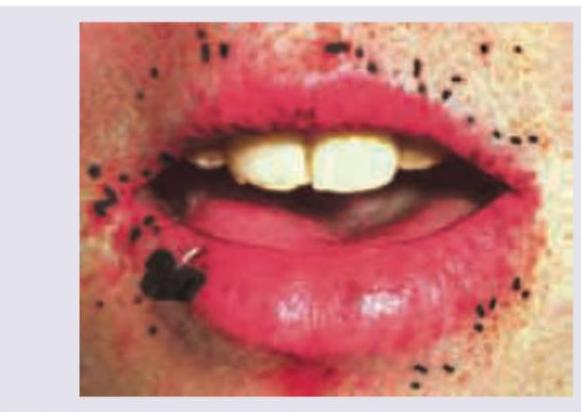
The child is having skeletal abnormality in arms and hands. Which possible heart disease is seen in this condition?
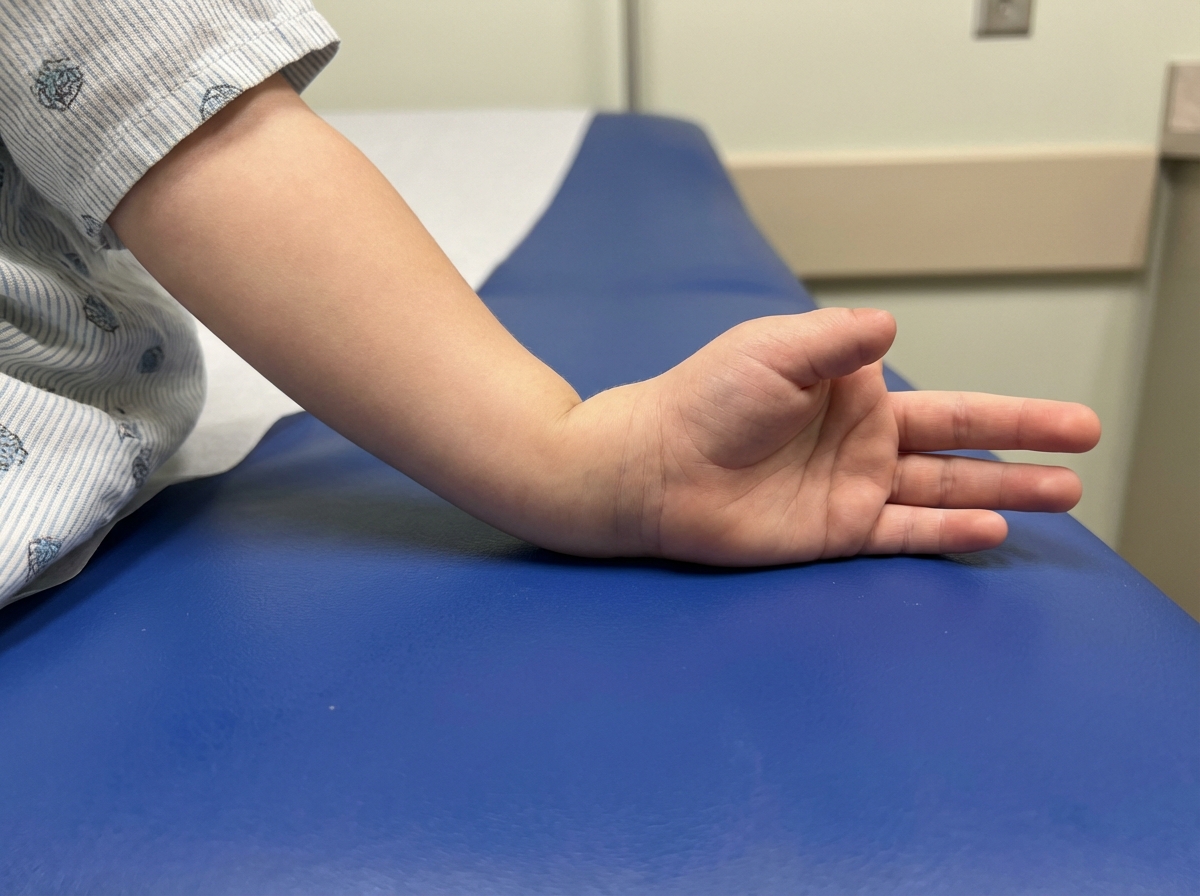
The image reveals hyper-extensible skin seen with Ehlers-Danlos syndrome. Which of the following is commonly seen in Marfan syndrome?
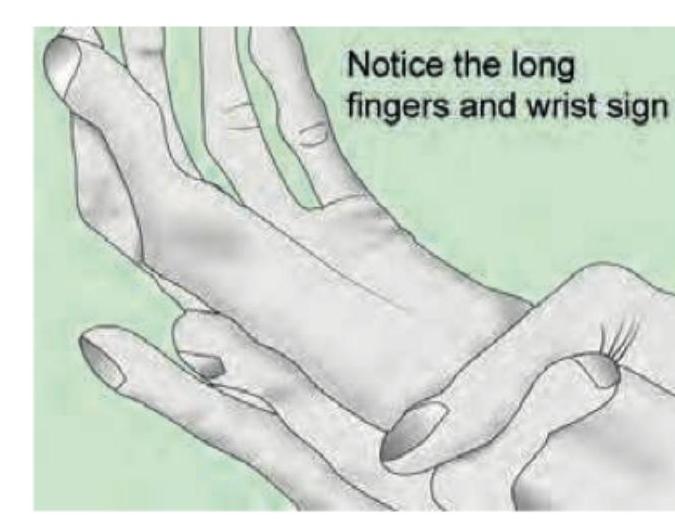
The image shows a tall patient with positive urine nitroprusside test diagnostic of Homocystinuria. Which of the following is true regarding Homocystinuria compared to Marfan's syndrome?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app