
Genetics and Disease — MCQs

On this page
A female child presents with virilization and hypertension with low plasma renin. What is the most likely diagnosis?
Which of the following statements is false regarding Menkes disease?
What is the most common cardiac defect in Turner syndrome?
Locus heterogeneity is a feature of which of the following?
All of the following diseases are associated with trinucleotide repeat sequences except?
What is the term for a single gene defect that causes multiple, seemingly unrelated problems?
Schwannoma of spinal nerve roots is typically seen in which condition?
What is the most sensitive investigation for cystic fibrosis?
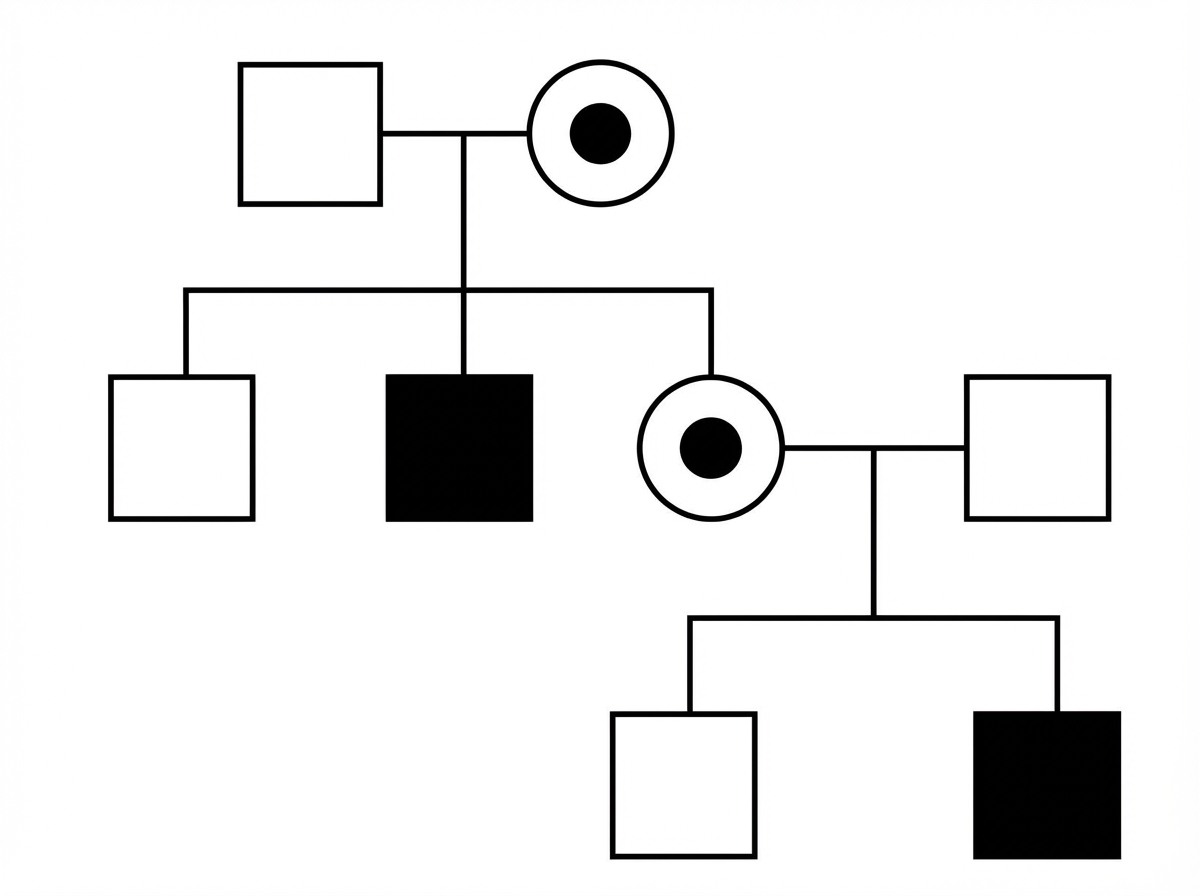
Study the following pedigree. What is the inheritance pattern of the disease in the family?
DNA topoisomerase 1 autoantibody is specific for which condition?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app