
Genetics and Disease — MCQs

On this page
Which of the following conditions is inherited in an autosomal dominant pattern?
Klinefelter syndrome is diagnosed by?
An albino girl marries a phenotypically normal boy. What are the chances of their having an affected child, and what are the chances of their children being carriers?
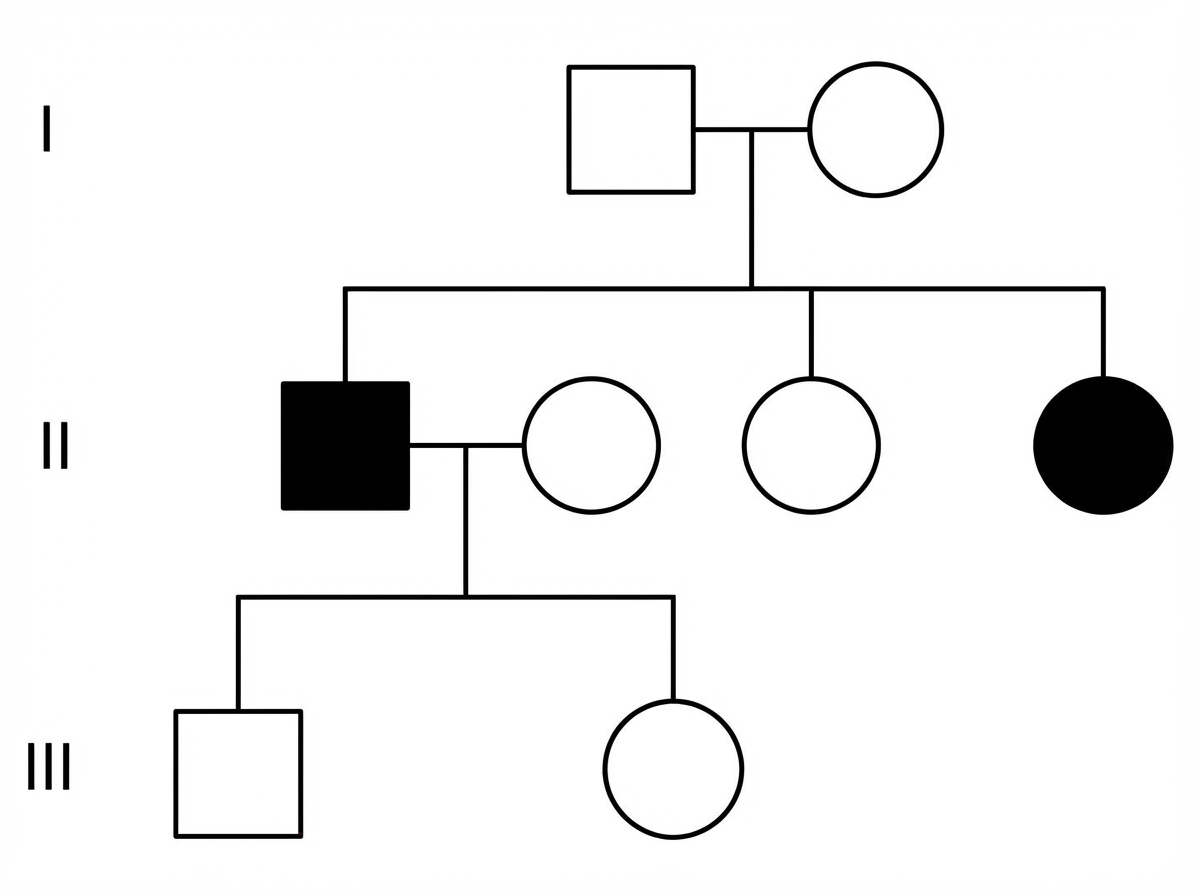
A pedigree chart shows the following pattern of inheritance. What is the most likely mode of inheritance?
Which of the following is an X-linked dominant condition?
Which enzyme levels are increased in Duchenne's muscular dystrophy?
A 32-year-old woman presents with a 2-day history of decreased vision and photopsia in her left eye. She is noted to be unusually tall with long limbs and slender fingers. Physical examination reveals pectus excavatum, kyphoscoliosis, a midsystolic click, and a pansystolic murmur. Ophthalmologic examination shows a retinal detachment and lens dislocation. The patient is at greatest risk for which of the following conditions?
All are manifestations of tuberous sclerosis, EXCEPT:
What is the commonest mode of inheritance of von Willebrand's disease?
A very tall, slender 16-year-old boy is referred for evaluation of an abnormal CXR. He reports no pulmonary or cardiac symptoms and feels well. On physical examination, he has long fingers, pectus excavatum, and a high arched palate. Which of the following is most likely to be seen on his CXR?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app