
Genetics and Disease — MCQs

On this page
Which of the following is not true in the case of myotonic dystrophy?
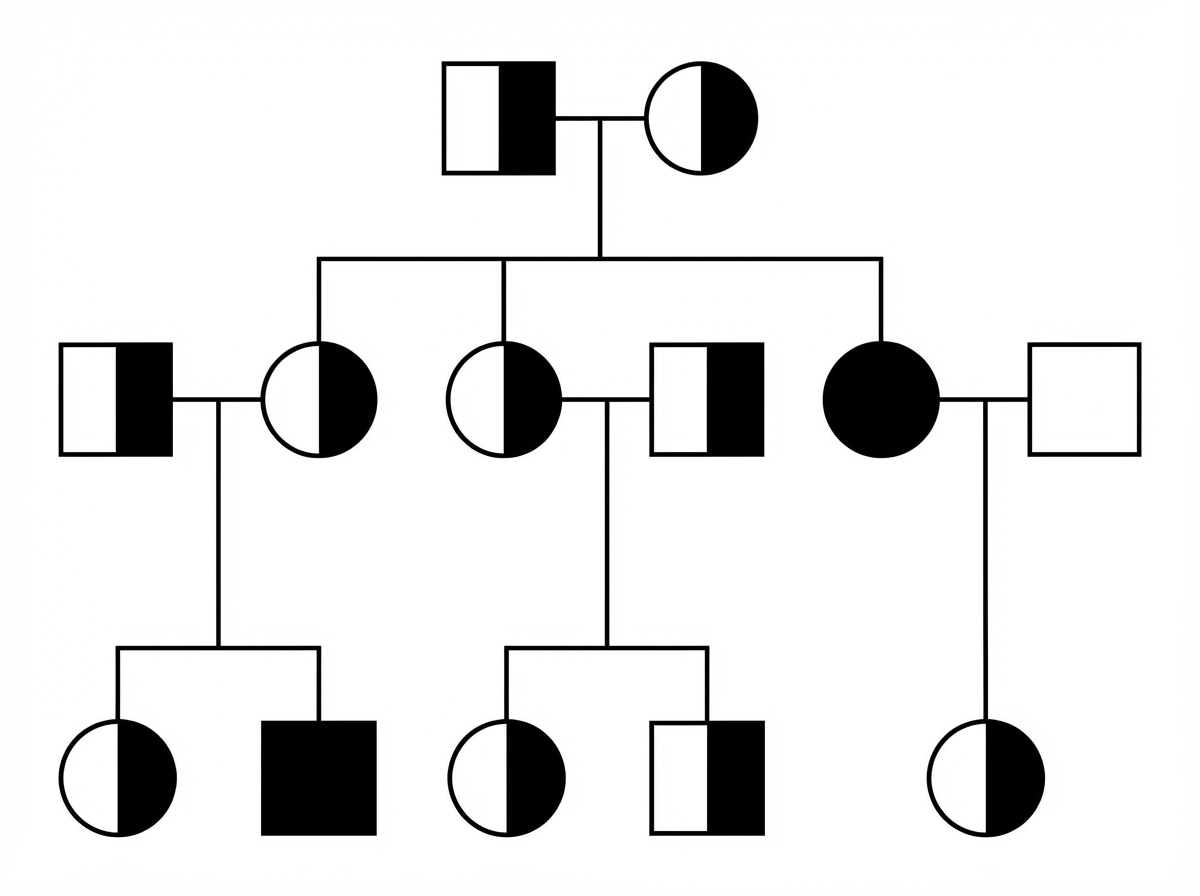
Examine the given pedigree chart. Which one of the following diseases is the most likely for this situation?
Myotonic dystrophy is inherited on which chromosome?
Unusual extensibility of the tongue is a characteristic feature of which of the following conditions?
Mental retardation is NOT a feature of which of the following mucopolysaccharidoses?
Duchenne's muscular dystrophy is inherited in which pattern?
Which condition has an autosomal dominant inheritance pattern?
Which single gene disorder does not follow Mendelian inheritance?
A 35-year-old nonsmoking male has been diagnosed with emphysema. His father died of emphysema at age 30, but he smoked. His father also had cirrhosis and recurrent pancreatitis but did not drink alcohol. Which one of the following inheritance patterns typifies this disease process?
Adult Polycystic kidney disease is inherited by which mode?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app