
Genetics and Disease — MCQs

On this page
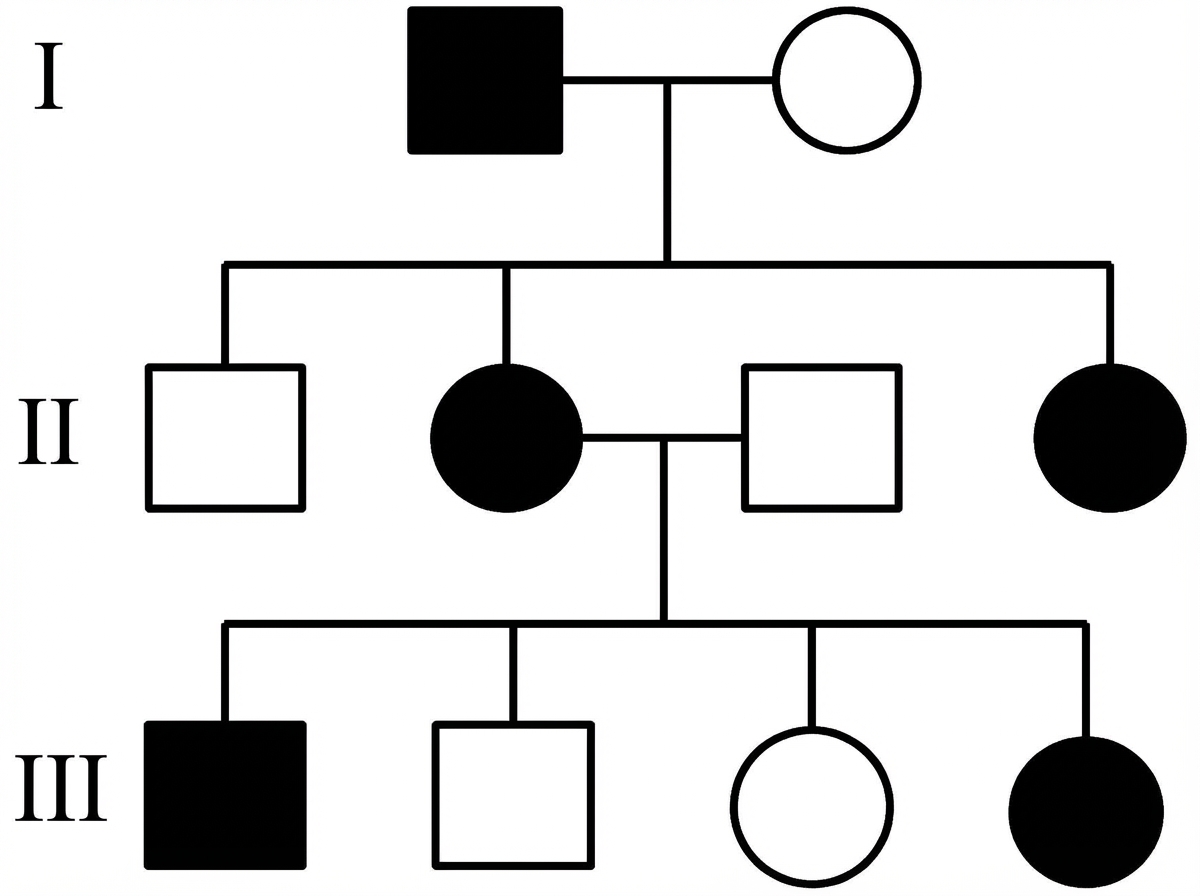
Which is the most likely inheritance pattern of the disease shown in the family pedigree?
Von Hippel-Lindau disease is associated with all the following except?
Phenotypic expression of a gene depending on the parent of origin is referred to as _______
Which of the following is the inheritance pattern of Incontinentia Pigmenti?
Mucociliary clearance is increased in cystic fibrosis by which of the following agents?
In patients with hypertrophic cardiomyopathy, in which gene are the maximum mutations found?
Which of the following is NOT an autosomal dominant disorder?
Which condition is Autosomal dominant?
Classic form of Alport syndrome is inherited as:
Which of the following statements about Hemochromatosis is true?
Practice by Chapter
Principles of Medical Genetics
Practice Questions
Genetic Testing and Counseling
Practice Questions
Single Gene Disorders
Practice Questions
Chromosomal Disorders
Practice Questions
Mitochondrial Diseases
Practice Questions
Pharmacogenomics
Practice Questions
Cancer Genetics
Practice Questions
Genetics of Common Diseases
Practice Questions
Epigenetics and Disease
Practice Questions
Genetic Basis of Developmental Disorders
Practice Questions
Ethical Issues in Medical Genetics
Practice Questions
Gene Therapy and Precision Medicine
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app