Back
USMLE Step 1 Biochemistry: High-Yield Topics, Pathways and Study Strategy (2026)
Master USMLE Step 1 biochemistry with high-yield topics, metabolic pathways, enzyme deficiencies, and proven study strategies. Complete guide to amino acids, vitamins, and clinical connections.

USMLE Step 1 Biochemistry: High-Yield Topics, Pathways and Study Strategy (2026)
You are probably staring at a 600-page biochemistry textbook right now, wondering how anyone memorizes every single enzyme in human metabolism. Here's the truth: they dont. Step 1 biochemistry isnt about memorizing every intermediate — its about understanding pattern recognition and clinical connections.
USMLE Step 1 has 280 questions. Roughly 25-35 of them are pure biochemistry, with another 15-20 mixing biochem into pathology or pharmacology vignettes. That means 50 questions could hinge on whether you truly understand amino acid disorders, enzyme kinetics, or why someone with Gaucher disease has a glucocerebrosidase deficiency.
The students who nail biochemistry dont just memorize pathways — they own the logic. When they see "intellectual disability + musty odor," they immediately think PKU → phenylalanine hydroxylase deficiency → tyrosine becomes essential. When they see competitive inhibition on a Lineweaver-Burk plot, they know Km increases but Vmax stays the same.
This guide breaks down the exact high-yield topics, pathway logic, and study strategies that separate 240+ scorers from everyone else.
Why Biochemistry Trips Up Most Students
Biochemistry feels overwhelming because medical school teaches it backwards. You learn glycolysis in September, TCA cycle in October, fatty acid oxidation in November — all as isolated units. By Step 1, you cant see how they connect.
Step 1 biochemistry questions test integration. They give you a patient with carnitine deficiency and ask why they cant oxidize long-chain fatty acids during fasting. Or they show you someone with B1 deficiency and expect you to know that pyruvate dehydrogenase (and α-ketoglutarate dehydrogenase) need thiamine pyrophosphate as a cofactor.
The fix is simple: learn pathways as connected systems, not isolated reactions. Start with the big picture, then drill down to enzyme deficiencies and clinical presentations.
High-Yield Biochemistry Topics for Step 1
1. Amino Acid Metabolism and Disorders
Core concept: Most amino acid disorders involve enzyme deficiencies that block normal degradation pathways, causing toxic buildup of substrates. High-yield disorders:
PKU (Phenylketonuria): Phenylalanine hydroxylase deficiency → phenylalanine buildup → intellectual disability, musty odor, fair skin. Treatment: low-phenylalanine diet, tyrosine becomes essential.
Maple syrup urine disease: Branched-chain α-keto acid dehydrogenase deficiency → leucine, isoleucine, valine buildup → sweet-smelling urine, encephalopathy
Homocystinuria: Cystathionine synthase deficiency → homocysteine buildup → lens dislocation (downward), intellectual disability, thrombosis
Alkaptonuria: Homogentisate oxidase deficiency → homogentisic acid buildup → dark urine, arthritis, dark connective tissue
The pattern here is enzyme name + clinical presentation + dietary management. Oncourse's spaced repetition flashcards automatically bring back these amino acid disorder cards right before you forget them — the SM-2 algorithm adapts based on whether you rate each card Easy, Good, Hard, or Forgot.
2. Urea Cycle Disorders
The urea cycle converts toxic ammonia to less toxic urea. Each enzyme deficiency causes hyperammonemia but with different clinical patterns.
Key enzymes (in order):
1. Carbamoyl phosphate synthetase I (CPS I)
2. Ornithine transcarbamylase (OTC) — X-linked, most common
3. Argininosuccinate synthetase
4. Argininosuccinate lyase
5. Arginase
Clinical pearls:
OTC deficiency is X-linked → affects males more severely
All cause hyperammonemia → neurologic symptoms, cerebral edema
Treatment: restrict protein, give sodium benzoate/phenylbutyrate to create alternative nitrogen disposal routes
Practice these disorders with targeted urea cycle MCQs to cement the enzyme sequence and clinical presentations.
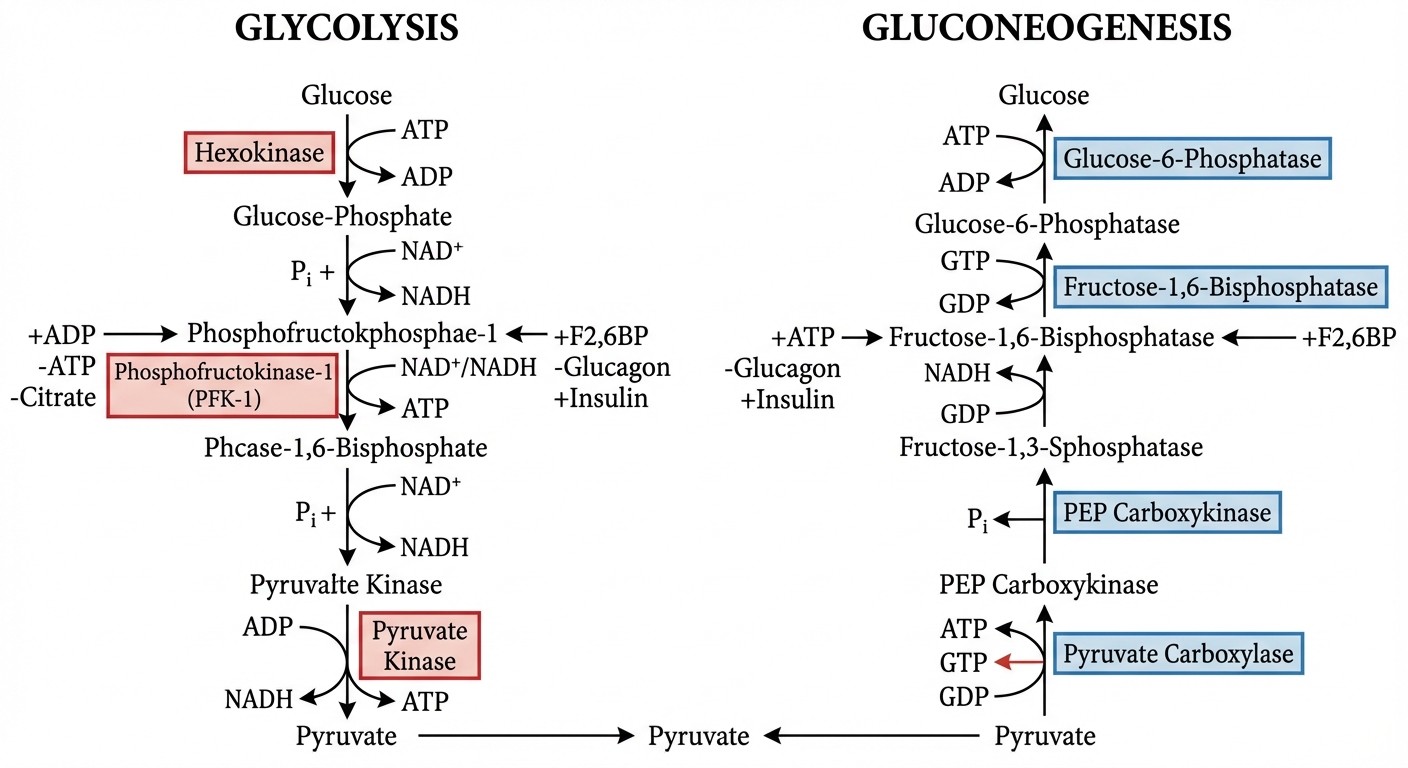
3. Glycolysis and Gluconeogenesis
Glycolysis regulation (know these 3 rate-limiting enzymes):
Hexokinase: inhibited by glucose-6-phosphate (product inhibition)
Phosphofructokinase-1 (PFK-1): key regulatory enzyme, inhibited by ATP and citrate, activated by AMP and ADP
Pyruvate kinase: inhibited by ATP, activated by glucose and insulin
Gluconeogenesis: essentially glycolysis in reverse, but uses different enzymes to bypass the irreversible steps. Key concept: occurs during fasting to maintain blood glucose. Clinical connection: PFK-1 deficiency (Tarui disease) causes exercise intolerance and hemolysis — muscle cant efficiently use glucose for energy.
4. TCA Cycle and Clinical Connections
The TCA cycle isnt just about memorizing intermediates — Step 1 tests specific clinical connections.
High-yield connections:
α-Ketoglutarate dehydrogenase: needs thiamine (B1), same as pyruvate dehydrogenase → B1 deficiency affects both
Succinyl-CoA: connects to heme synthesis and ketone body metabolism
Oxaloacetate: depleted during gluconeogenesis → can slow TCA cycle during fasting
Clinical pearl: Arsenic poisoning inhibits pyruvate dehydrogenase and α-ketoglutarate dehydrogenase by binding to lipoic acid cofactors.
5. Fatty Acid Metabolism
β-Oxidation essentials:
Requires carnitine shuttle to get fatty acids into mitochondria
Each cycle produces 1 acetyl-CoA, 1 FADH2, 1 NADH
Odd-chain fatty acids: final 3-carbon fragment becomes propionyl-CoA → methylmalonyl-CoA → succinyl-CoA (needs B12)
Ketone bodies: produced when acetyl-CoA exceeds TCA cycle capacity (during fasting or diabetes)
Liver makes them (acetoacetate, β-hydroxybutyrate)
Peripheral tissues use them for energy
Brain can adapt to use ketones during prolonged fasting
For fatty acid oxidation disorders and ketone body metabolism, Oncourse's adaptive daily plans automatically balance new concept learning with MCQ practice based on your study timeline and progress.
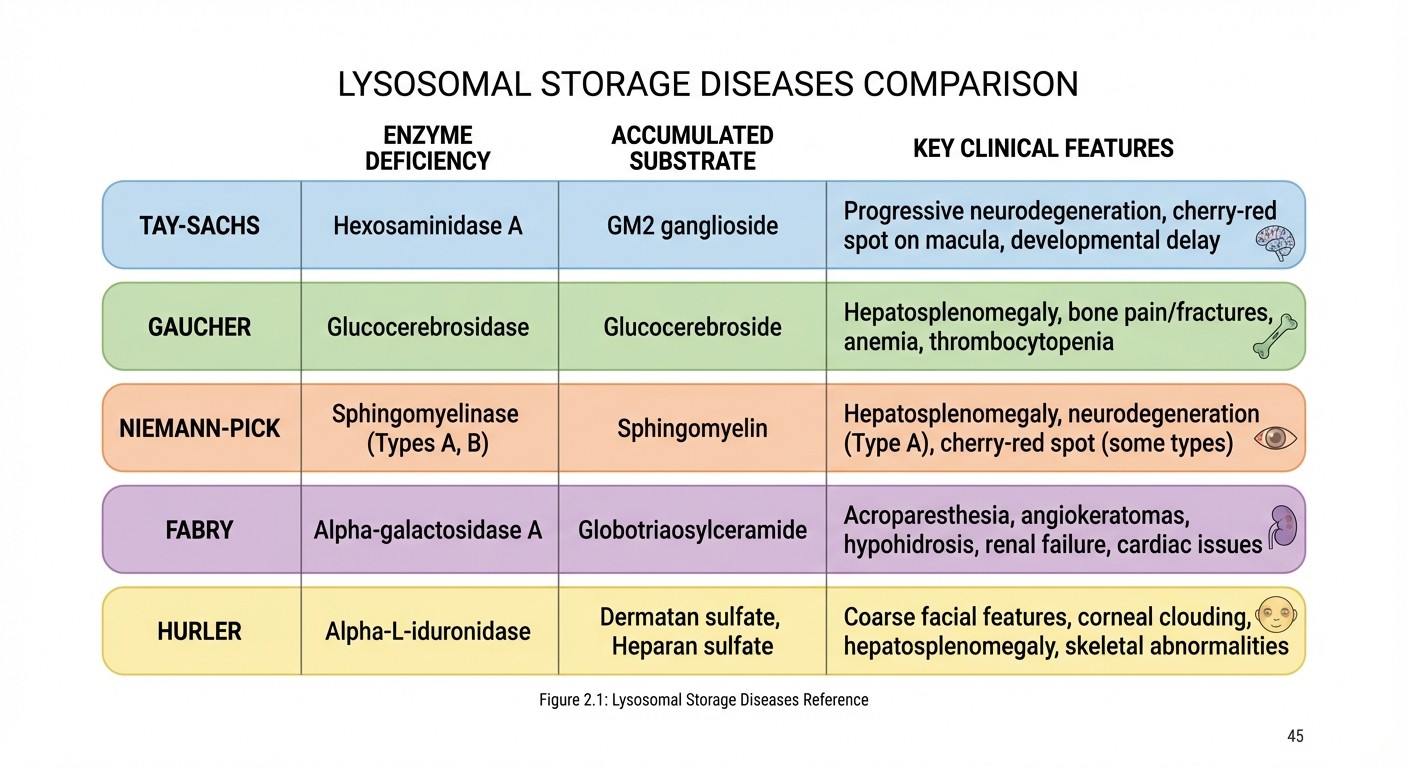
6. Lysosomal Storage Diseases
These are enzyme deficiencies affecting lysosomal breakdown of complex molecules. Step 1 loves testing the enzyme-substrate combinations.
Sphingolipidoses (know enzyme + accumulated substrate):
Tay-Sachs: hexosaminidase A deficiency → GM2 ganglioside accumulation → cherry-red spot, intellectual disability
Gaucher: glucocerebrosidase deficiency → glucocerebroside accumulation → hepatosplenomegaly, bone pain
Niemann-Pick: sphingomyelinase deficiency → sphingomyelin accumulation → hepatosplenomegaly, cherry-red spot
Fabry: α-galactosidase deficiency → ceramide trihexoside accumulation → peripheral neuropathy, angiokeratomas
Mucopolysaccharidoses:
Hurler syndrome: α-L-iduronidase deficiency → heparan sulfate and dermatan sulfate accumulation → coarse facial features, intellectual disability
The pattern is always the same: enzyme deficiency → substrate accumulation → organ dysfunction. Master this with lysosomal storage disease lessons that break down each disease systematically.
7. Purine and Pyrimidine Metabolism
De novo vs. salvage pathways:
De novo: builds purines/pyrimidines from scratch (expensive, uses lots of energy)
Salvage: recycles existing bases (cheaper, more efficient)
Key enzymes:
HGPRT (Hypoxanthine-guanine phosphoribosyltransferase): salvage pathway for purines
Adenosine deaminase (ADA): breaks down adenosine to inosine
Clinical connections:
Lesch-Nyhan syndrome: HGPRT deficiency → cant salvage purines → overproduction via de novo pathway → hyperuricemia, self-mutilation, intellectual disability
ADA deficiency: adenosine buildup toxic to lymphocytes → severe combined immunodeficiency (SCID)
8. Vitamins and Cofactor Deficiencies
Fat-soluble vitamins (A, D, E, K):
Vitamin A: retinal (vision), retinoic acid (gene transcription) → deficiency causes night blindness, xerophthalmia
Vitamin D: calcium homeostasis → deficiency causes rickets (children), osteomalacia (adults)
Vitamin E: antioxidant → deficiency causes hemolysis, peripheral neuropathy
Vitamin K: cofactor for clotting factors II, VII, IX, X → deficiency causes bleeding
Water-soluble B vitamins (know the enzyme cofactor):
B1 (Thiamine): TPP cofactor for pyruvate dehydrogenase, α-ketoglutarate dehydrogenase → deficiency causes beriberi, Wernicke-Korsakoff
B2 (Riboflavin): FAD/FMN cofactor → deficiency causes angular cheilitis, glossitis
B3 (Niacin): NAD+/NADH cofactor → deficiency causes pellagra (4 Ds: dermatitis, diarrhea, dementia, death)
B6 (Pyridoxine): PLP cofactor for amino acid metabolism → deficiency causes peripheral neuropathy, seizures
B12 (Cobalamin): methylmalonyl-CoA mutase cofactor → deficiency causes megaloblastic anemia, subacute combined degeneration
Folate: one-carbon metabolism → deficiency causes megaloblastic anemia, neural tube defects
9. Enzyme Kinetics
Step 1 tests Michaelis-Menten kinetics more than students expect. Know these concepts cold:
Michaelis-Menten equation: V = (Vmax × [S]) / (Km + [S])
Km: substrate concentration at half-maximal velocity (measures enzyme affinity)
Vmax: maximum reaction velocity (measures enzyme amount/activity)
Inhibition types:
Competitive: inhibitor competes with substrate for active site → Km increases, Vmax unchanged
Non-competitive: inhibitor binds allosteric site → Vmax decreases, Km unchanged
Uncompetitive: inhibitor binds enzyme-substrate complex → both Km and Vmax decrease
Lineweaver-Burk plots: double reciprocal plot (1/V vs. 1/[S]) makes inhibition types easier to distinguish.
Whenever you encounter enzyme kinetics MCQs, Oncourse's AI-powered explanations break down why each answer choice is right or wrong, helping you move from rote memorization to genuine mechanistic understanding of Km and Vmax changes.
10. Molecular Biology Essentials
DNA replication:
Leading strand: synthesized continuously 5' → 3'
Lagging strand: synthesized discontinuously as Okazaki fragments
Key enzymes: DNA polymerase III (prokaryotes), DNA polymerase α, δ, ε (eukaryotes)
DNA repair mechanisms:
Mismatch repair: fixes base mispairing
Nucleotide excision repair: fixes thymine dimers, bulky adducts
Base excision repair: fixes single altered bases
Transcription and translation inhibitors (antibiotic mechanisms):
Rifampin: blocks RNA polymerase
Actinomycin D: intercalates DNA, blocks transcription
Chloramphenicol: blocks peptidyl transferase (50S ribosome)
Streptomycin: blocks initiation, causes misreading (30S ribosome)
Step 1 Biochemistry Study Strategy
Phase 1: Build the Foundation (Weeks 1-2)
Start with pathway logic before diving into details. Draw major metabolic pathways from memory:
Glycolysis (10 steps)
TCA cycle (8 steps)
β-oxidation (4-step cycle)
Urea cycle (5 enzymes)
Dont just read — actively reconstruct these pathways. When you can draw glycolysis with all regulatory points and enzyme names, you own it.
Use biochemistry lessons to build this foundation systematically. The lessons connect individual pathways into integrated metabolic networks — exactly how Step 1 tests them.
Phase 2: Clinical Integration (Weeks 3-4)
Now connect pathways to disease states. For every enzyme deficiency disorder, know:
1. Enzyme affected
2. Substrate that accumulates
3. Clinical presentation
4. Treatment approach
This is where spaced repetition becomes crucial. Biochemistry has tons of details that fade quickly without reinforcement. Set up your flashcard reviews to hit enzyme deficiencies, vitamin cofactors, and clinical connections daily.
Phase 3: MCQ Pattern Recognition (Weeks 5-6)
Biochemistry MCQs follow predictable patterns:
Enzyme deficiency vignettes: patient presentation → identify affected enzyme → predict accumulated substrate
Metabolic integration: fasting/fed state → which pathways are active → regulatory enzyme effects
Vitamin deficiency: clinical symptoms → identify deficient vitamin → know the affected enzyme/pathway
Practice MCQs by topic blocks first (pure glycolysis questions, pure amino acid disorder questions), then switch to mixed biochemistry sets. This builds pattern recognition without overwhelming you.
Phase 4: Weak Spot Elimination (Final 2 weeks)
Review your missed questions and identify gaps. Common weak spots:
Confusing lysosomal storage diseases (memorize the enzyme-substrate pairs)
Forgetting odd-chain fatty acid metabolism (propionyl-CoA → methylmalonyl-CoA → succinyl-CoA, needs B12)
Missing enzyme kinetics concepts (competitive vs. non-competitive inhibition effects)
Not knowing vitamin cofactors (which B vitamin is needed for specific enzyme reactions)
Drill these specific topics until they become automatic.
Common Biochemistry Pitfalls
Pitfall 1: Passive Reading Instead of Active Recall
Reading biochemistry textbooks feels productive but doesnt stick. Your brain recognizes the pathway diagrams but cant reconstruct them independently.
Fix: Close the book and draw pathways from memory. If you cant complete glycolysis without peeking, you dont know it yet.
Pitfall 2: Ignoring Enzyme Kinetics
Many students skip enzyme kinetics because it feels "too theoretical." Bad move — Step 1 regularly tests Km/Vmax concepts and inhibition types.
Fix: Learn the Michaelis-Menten equation and practice Lineweaver-Burk plot interpretation. Know that competitive inhibition increases Km but leaves Vmax unchanged.
Pitfall 3: Memorizing Without Understanding Logic
You can memorize that PKU involves phenylalanine hydroxylase deficiency, but do you understand why tyrosine becomes an essential amino acid? The logic matters more than rote facts.
Fix: For every enzyme deficiency, trace the pathway. PKU blocks phenylalanine → tyrosine conversion, so dietary tyrosine becomes essential since the body cant make it.
Pitfall 4: Confusing Lysosomal Storage Diseases
Tay-Sachs, Gaucher, Niemann-Pick, and Fabry disease blur together because they all involve sphingolipid accumulation.
Fix: Make a table with enzyme, accumulated substrate, and key clinical features. Tay-Sachs = hexosaminidase A + GM2 ganglioside + cherry-red spot. Gaucher = glucocerebrosidase + glucocerebroside + hepatosplenomegaly.
Pitfall 5: Skipping Vitamin Cofactor Connections
Step 1 loves asking about vitamin deficiencies through enzyme dysfunction. B1 deficiency affects pyruvate dehydrogenase AND α-ketoglutarate dehydrogenase because both need thiamine pyrophosphate.
Fix: For every B vitamin, memorize the specific enzyme it affects as a cofactor. B12 is needed for methylmalonyl-CoA mutase — thats why B12 deficiency causes methylmalonic aciduria.
Advanced Study Tips
Use Mnemonics Strategically
Some biochemistry facts require brute force memorization. Use mnemonics for high-yield lists:
Fat-soluble vitamins: "ADEK" (A, D, E, K) Branched-chain amino acids: "LIVe" (Leucine, Isoleucine, Valine) Aromatic amino acids: "FTW" (Phenylalanine, Tyrosine, Tryptophan)
Oncourse's Synapses feature creates visual memory associations for complex biochemistry topics — like connecting the maple syrup smell of MSUD with branched-chain amino acid buildup through memorable imagery.
Connect to Pharmacology
Biochemistry and pharmacology overlap heavily on Step 1. Many drug mechanisms involve blocking specific enzymes:
Statins: block HMG-CoA reductase (cholesterol synthesis)
Allopurinol: blocks xanthine oxidase (purine metabolism)
Methotrexate: blocks dihydrofolate reductase (folate metabolism)
Understanding the biochemical pathway makes the drug mechanism obvious.
Review Missed Questions Immediately
When you miss a biochemistry MCQ, dont just read the explanation — trace the entire pathway. If you missed a question about homocystinuria, review the entire methionine degradation pathway, not just the single enzyme deficiency.
This deeper review prevents related mistakes and builds comprehensive understanding.
Sample Study Schedule
Week 1: Glycolysis, gluconeogenesis, TCA cycle (pathway memorization + regulation) Week 2: Fatty acid metabolism, ketone bodies, amino acid metabolism basics Week 3: Amino acid disorders (PKU, MSUD, homocystinuria, alkaptonuria), urea cycle Week 4: Lysosomal storage diseases, purine/pyrimidine metabolism Week 5: Vitamins and cofactor deficiencies, enzyme kinetics Week 6: Molecular biology, mixed MCQ practice Final week: Review missed questions, drill weak spots
Spend 2-3 hours daily: 1 hour new content, 1 hour flashcard review, 1 hour MCQ practice.
Frequently Asked Questions
How much detail should I know for each metabolic pathway?
Focus on rate-limiting enzymes, regulatory points, and clinical connections. You dont need every intermediate for glycolysis, but you must know that PFK-1 is the key regulatory enzyme inhibited by ATP and citrate.
Should I memorize all the lysosomal storage diseases?
Yes, but focus on the high-yield ones: Tay-Sachs, Gaucher, Niemann-Pick, Fabry, and Hurler. Know the enzyme deficiency, accumulated substrate, and 2-3 key clinical features for each.
How important is enzyme kinetics for Step 1?
More important than most students realize. Expect 2-3 questions on Michaelis-Menten kinetics, competitive vs. non-competitive inhibition, and Lineweaver-Burk plots. Learn the concepts — dont just memorize formulas.
What's the best way to study amino acid disorders?
Group them by pathway: aromatic amino acid disorders (PKU, tyrosinemia), branched-chain disorders (MSUD), sulfur amino acid disorders (homocystinuria), etc. This creates logical connections instead of random memorization.
Should I use multiple biochemistry resources?
Pick one primary resource (textbook or online course) and supplement with MCQ practice. Jumping between multiple textbooks creates confusion because they organize topics differently. Consistency beats variety for biochemistry.
How do I connect biochemistry to other subjects?
Look for overlap: biochemistry enzyme deficiencies create pathology (PKU → intellectual disability), pharmacology targets biochemical pathways (statins → HMG-CoA reductase), and genetics explains inheritance patterns (X-linked OTC deficiency). Integration is key for Step 1 success.
Master Biochemistry, Master Step 1
USMLE Step 1 biochemistry isnt about memorizing every enzyme — its about pattern recognition and clinical integration. When you see hyperammonemia in a newborn, think urea cycle disorder. When you see competitive inhibition on a kinetics plot, know that Km increases but Vmax stays constant.
The students who excel dont just know biochemistry — they think biochemically. They see pathway connections, recognize clinical presentations, and understand why specific enzyme deficiencies cause predictable symptoms.
Build your foundation with pathway logic, drill clinical connections through spaced repetition, and practice MCQ pattern recognition until biochemical thinking becomes automatic.
Prepare smarter with Oncourse AI — adaptive MCQs, spaced repetition, and AI explanations built for USMLE Step 1. Download free on Android and iOS.