
Renal Pathology — MCQs

On this page
In a case of glomerulonephritis (GN), C3 is normal in all the following except?
Chromosomes associated with autosomal dominant polycystic kidney disease (ADPKD).
Bellini duct cancer is seen in which of the following?
Loss of foot processes seen on electron microscopy of renal biopsy is a classical feature in which of the following?
Which of the following is associated with pauci-immune glomerulonephritis?
Immunofluorescence staining pattern from a kidney biopsy from a 35-year-old patient presenting with proteinuria has been shown below. What is the most probable cause?
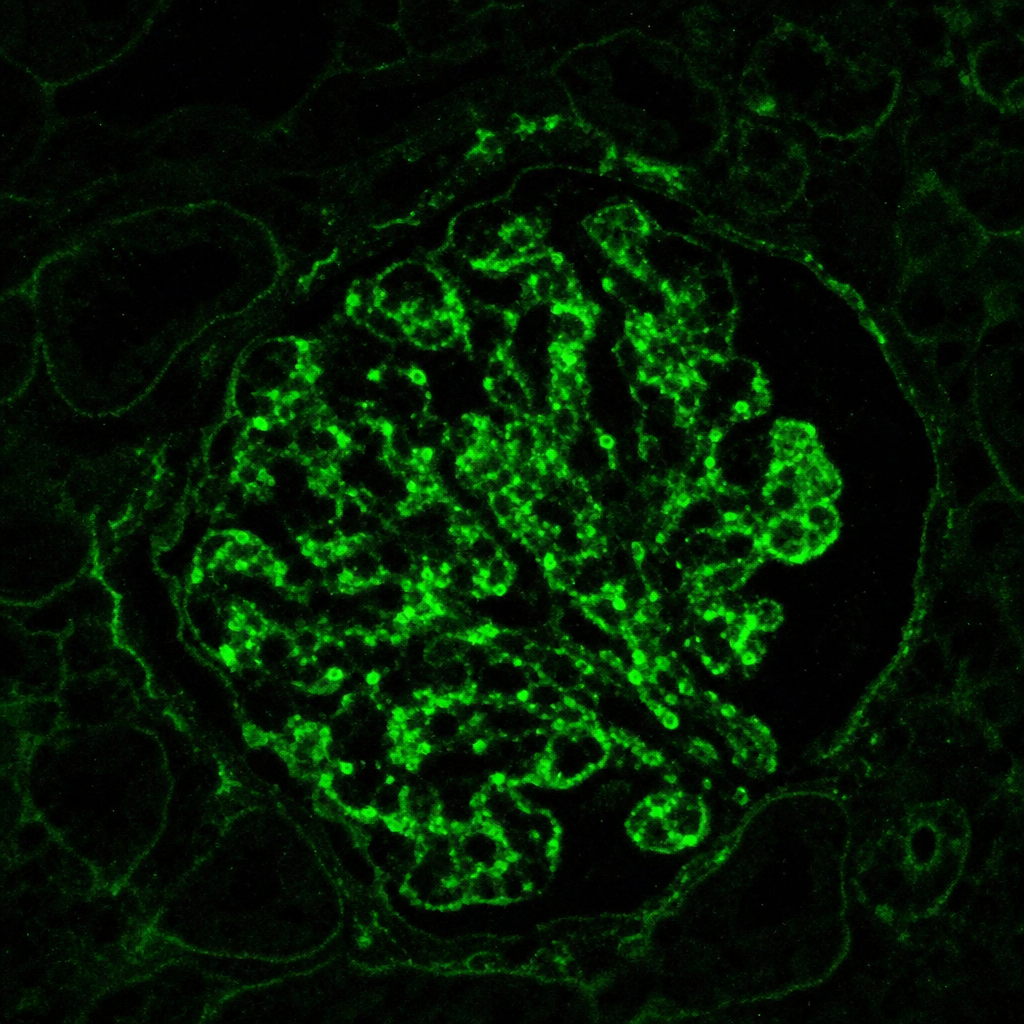
Immunofluorescence staining pattern from a kidney biopsy from a 35-year-old patient presenting with proteinuria has been shown below. What is the most probable cause?
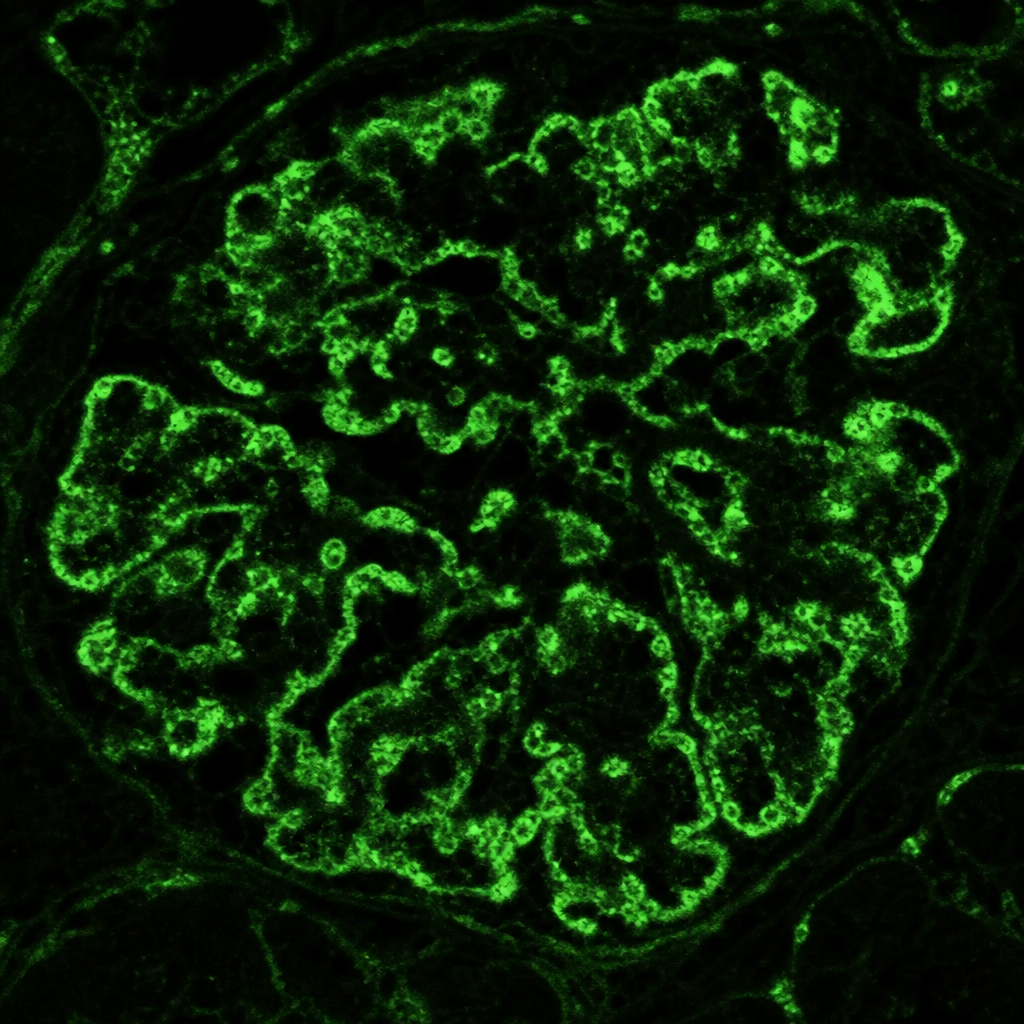
Renal biopsy shows 'spike and dome' appearance on silver methenamine stain. Immunofluorescence shows granular IgG deposits. Diagnosis?
Feature NOT seen in acute rejection of transplanted kidney?
Most specific test for diagnosing Fabry disease on kidney biopsy?
Practice by Chapter
Congenital Anomalies of the Kidney
Practice Questions
Glomerular Diseases
Practice Questions
Tubular and Interstitial Diseases
Practice Questions
Vascular Diseases of the Kidney
Practice Questions
Cystic Diseases of the Kidney
Practice Questions
Urinary Tract Obstruction and Stones
Practice Questions
Renal Tumors
Practice Questions
Kidney in Systemic Diseases
Practice Questions
Renal Transplantation Pathology
Practice Questions
Urinary Tract Infections
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app