
Renal Pathology — MCQs

On this page
In renal disease, why does albumin appear first in the urine?
What are the characteristic features of glomerular hematuria?
What is the most specific histological lesion in diabetic nephropathy?
Which chromosomal abnormality is associated with Chromophilic Renal Cell Carcinoma?
A 33-year-old woman presents with a one-week history of increasing lethargy and decreased urine output. Laboratory studies reveal a serum creatinine of 4.3 mg/dL and blood urea nitrogen of 40 mg/dL. A renal biopsy is performed and examined using electron microscopy. Which of the following morphologic cellular changes most likely suggests a diagnosis of acute tubular necrosis?
Which of the following is NOT a feature of familial hemolytic uremic syndromes?
What is the most common infectious agent associated with chronic pyelonephritis?
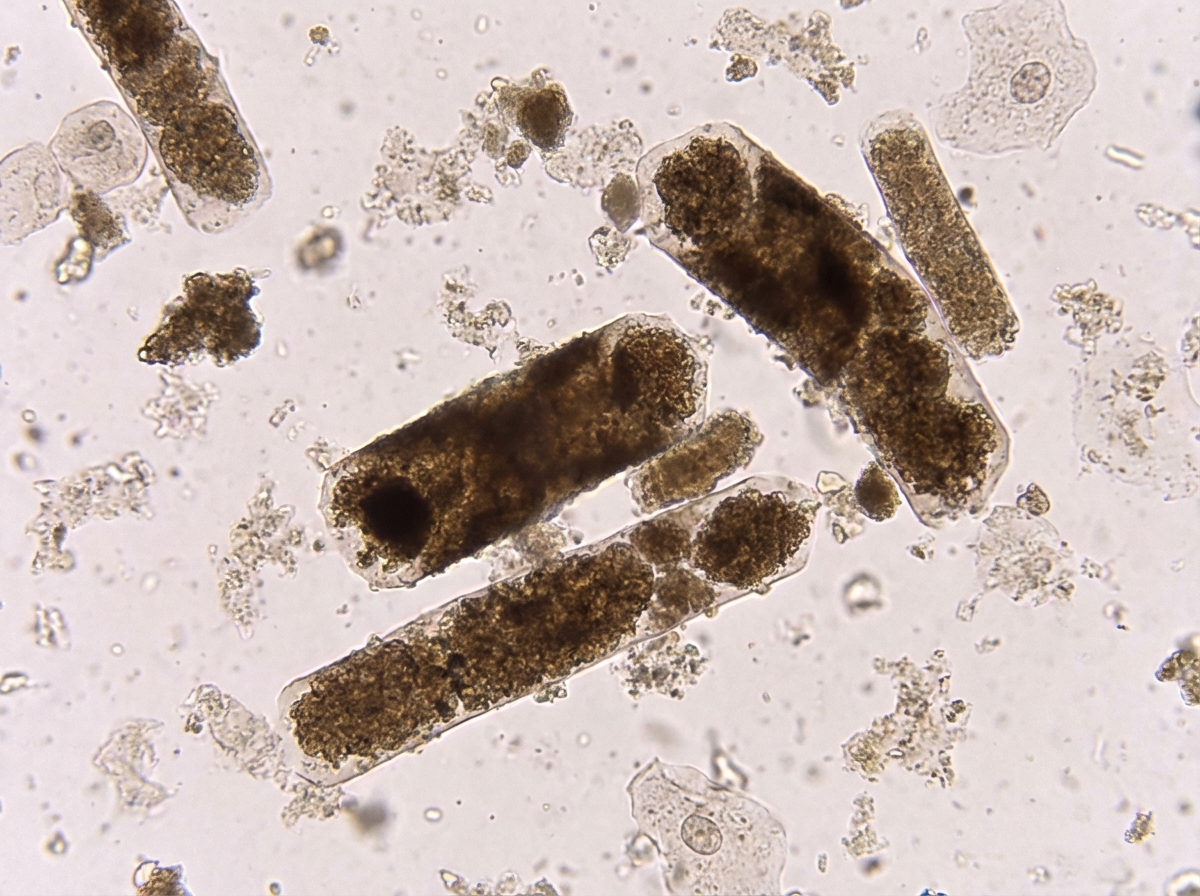
Which of the following conditions is characterized by the shown urine microscopy finding?
What is the most common cause of renal tumors in adults?
Which condition is characterized by hematuria with dysmorphic RBCs?
Practice by Chapter
Congenital Anomalies of the Kidney
Practice Questions
Glomerular Diseases
Practice Questions
Tubular and Interstitial Diseases
Practice Questions
Vascular Diseases of the Kidney
Practice Questions
Cystic Diseases of the Kidney
Practice Questions
Urinary Tract Obstruction and Stones
Practice Questions
Renal Tumors
Practice Questions
Kidney in Systemic Diseases
Practice Questions
Renal Transplantation Pathology
Practice Questions
Urinary Tract Infections
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app