
Renal Pathology — MCQs

On this page
Which of the following is NOT a feature of Minimal Change Disease?
A 4-year-old girl presents with swelling of the legs and ankles. Physical examination reveals pitting edema of the lower extremities. Urinalysis shows 2+ proteinuria. The urinary sediment contains no inflammatory cells or red blood cells. Serum levels of BUN and creatinine are normal. The patient recovers completely after a course of corticosteroids. Which of the following pathologic findings might be expected in the urine prior to treatment with corticosteroids?
Nephrotic syndrome is characterized by
All are features of hemolytic uremic syndrome, EXCEPT?
Minimal-change nephropathy is characterized by which of the following?
A 7-year-old boy has become less active over the past 10 days. On physical examination, the boy has facial puffiness. Urinalysis shows no blood, glucose, or ketones, and microscopic examination shows no casts or crystals. The serum creatinine level is normal. A 24-hour urine collection yields 3.8 g of protein. He improves after corticosteroid therapy. He has two more episodes of proteinuria over the next 4 years, both of which respond to corticosteroid therapy. What is the most likely mechanism causing his disease?
A 28-year-old man presents with a large amount of blood and protein in his urine. He has had sensorineural hearing loss since his teen years and anterior lenticonus. The physician suspects a genetic disorder that may lead to eventual kidney failure. If this is the case, the patient most likely has a mutation in which one of the following proteins?
Silver stain performed on renal biopsy shows characteristic findings. Immunostaining for Anti-PLA2R is positive. Based on the given findings, what is the likely diagnosis?
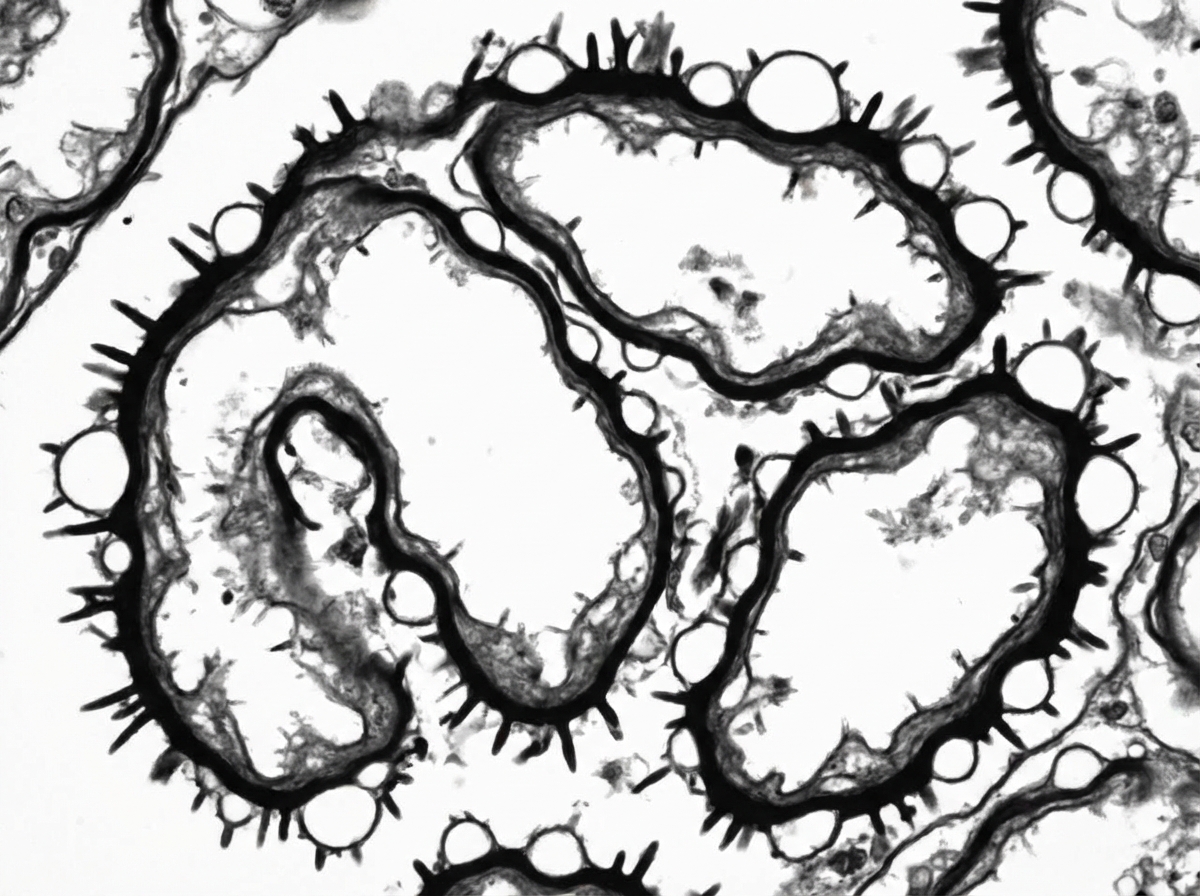
Rapidly progressive glomerulonephritis can be associated with all of the following, except?
Dysmorphic RBCs are seen in which of the following conditions?
Practice by Chapter
Congenital Anomalies of the Kidney
Practice Questions
Glomerular Diseases
Practice Questions
Tubular and Interstitial Diseases
Practice Questions
Vascular Diseases of the Kidney
Practice Questions
Cystic Diseases of the Kidney
Practice Questions
Urinary Tract Obstruction and Stones
Practice Questions
Renal Tumors
Practice Questions
Kidney in Systemic Diseases
Practice Questions
Renal Transplantation Pathology
Practice Questions
Urinary Tract Infections
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app