
Renal Pathology — MCQs

On this page
A 12-year-old presents with hypertension and hematuria. Serum C3 is raised. Renal biopsy shows subepithelial hump deposits on EM. What is the most likely diagnosis?
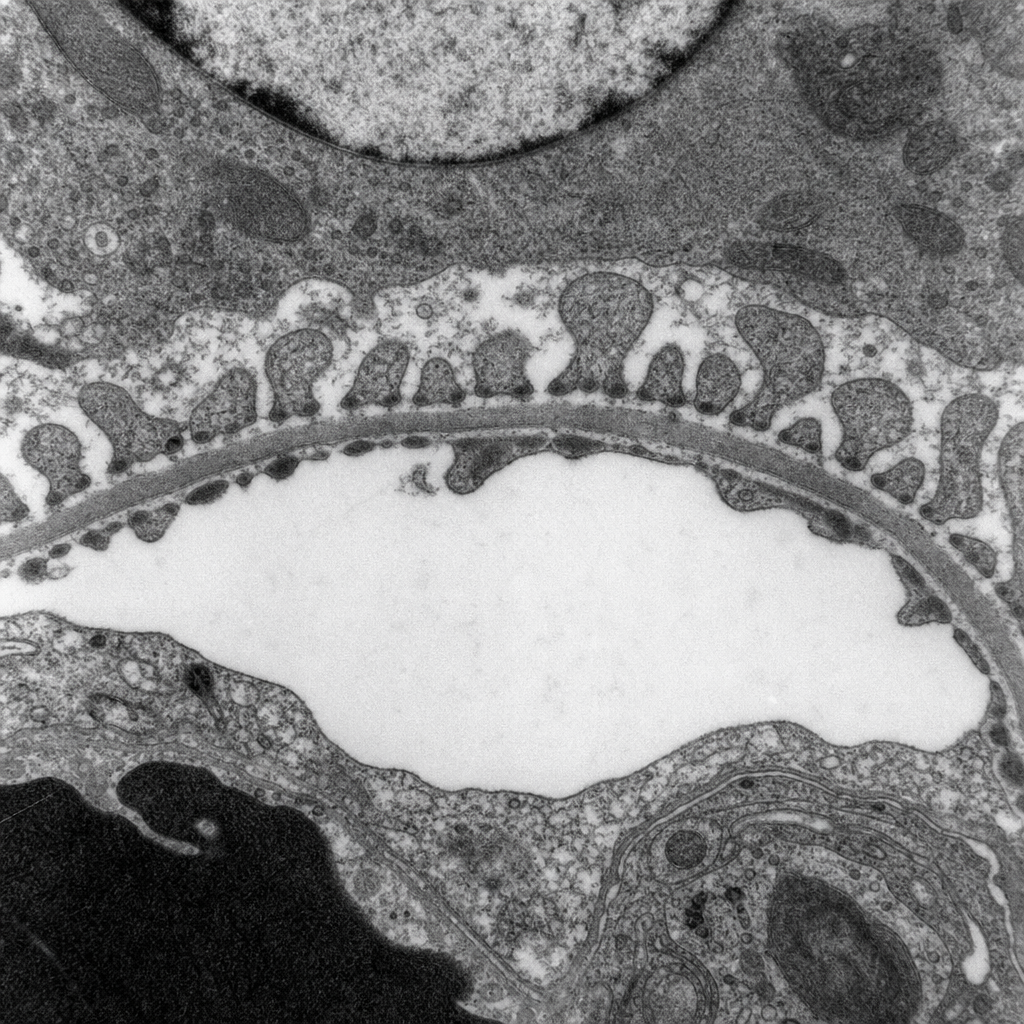
A 28-year-old woman presents with frothy urine, periorbital puffiness, and a serum albumin of 1.8 g/dL. Urinalysis reveals 4+ proteinuria with no haematuria. A renal biopsy is performed and the specimen is examined under electron microscopy (Image 1). The massive proteinuria in this condition results from dysfunction of which cell type?
Which chromosome is associated with Autosomal Dominant Polycystic Kidney Disease (ADPKD)?
What is the first pathological change apparent in Nephrotic syndrome?
Which nephritogenic antigen is detected in subepithelial humps of post-streptococcal glomerulonephritis (PSGN)?
Maximum 'Endocapillary Proliferation' is seen in which of the following conditions?
Squamous cell carcinoma of the urinary bladder is associated with which of the following?
Which of the following conditions can cause rapidly progressive glomerulonephritis?
What is the diagnostic urinary finding in pyelonephritis?
A 28-year-old male with AIDS presents with moderate proteinuria and hypertension. Histologic sections of the kidney reveal the combination of normal-appearing glomeruli and occasional glomeruli that have deposits of hyaline material. No increased cellularity or necrosis is noted in the abnormal glomeruli. Additionally, there is cystic dilation of the renal tubules, some of which are filled with proteinaceous material. Electron microscopy reveals focal fusion of podocytes, and immunofluorescence examination finds granular IgM/C3 deposits. What is the best diagnosis for this renal abnormality?
Practice by Chapter
Congenital Anomalies of the Kidney
Practice Questions
Glomerular Diseases
Practice Questions
Tubular and Interstitial Diseases
Practice Questions
Vascular Diseases of the Kidney
Practice Questions
Cystic Diseases of the Kidney
Practice Questions
Urinary Tract Obstruction and Stones
Practice Questions
Renal Tumors
Practice Questions
Kidney in Systemic Diseases
Practice Questions
Renal Transplantation Pathology
Practice Questions
Urinary Tract Infections
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app