
Neuropathology — MCQs

On this page
Group atrophy of muscle fibers denotes:
Cerebellar hemangioblastoma and retinal tumors are seen in which of the following conditions?
Which is the most common type of brain tumor?
Which tumor has the best prognosis?
Which of the following structures is most prone to hypoxic injury?
Elongated filaments in Pick's disease consist of?
Which of the following is NOT a neoplasm that patients with neurofibromatosis are prone to develop?
Onion bulb appearance on nerve biopsy is seen in which of the following conditions?
In the provided histopathology image of a schwannoma, what does the arrow-marked lesion represent?
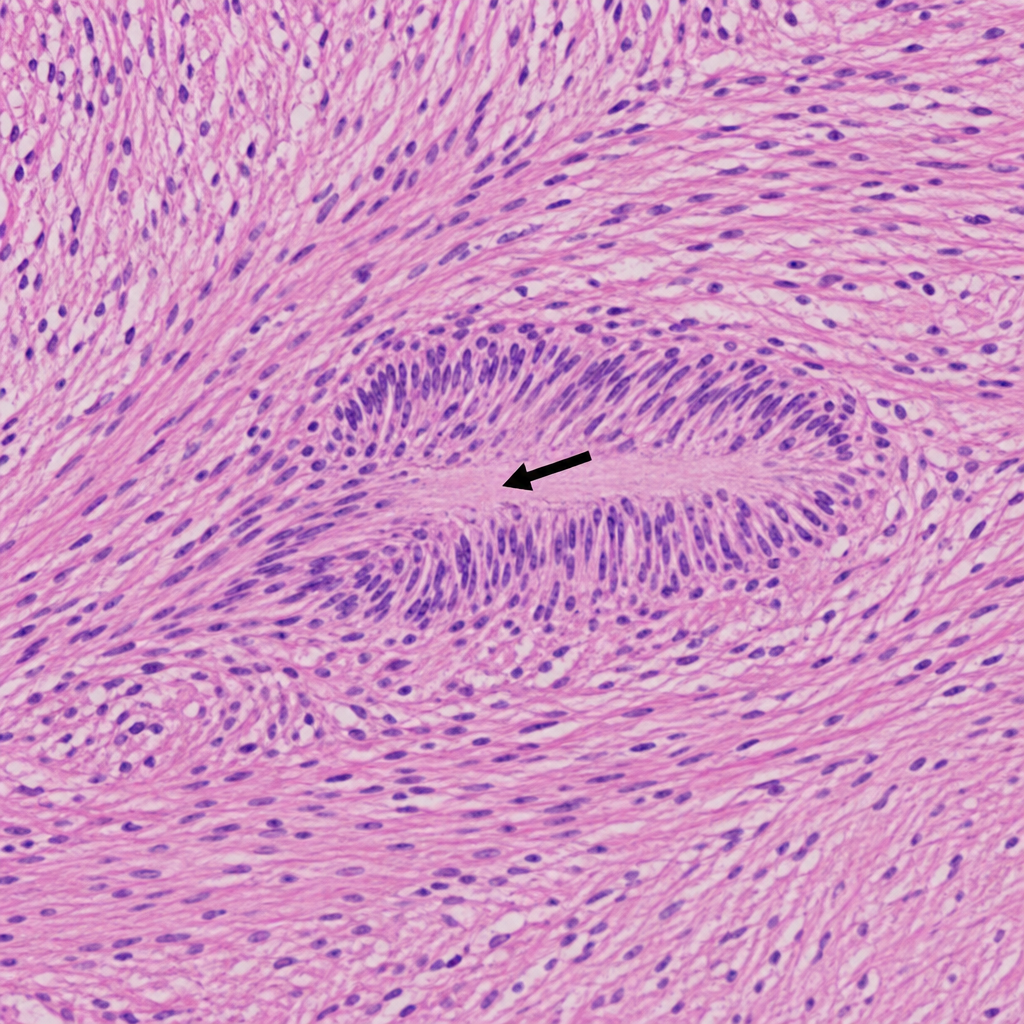
Which of the following conditions is characterized by cafe-au-lait spots, non-encapsulation, and potential for malignant transformation?
Practice by Chapter
Cellular Pathology of the Nervous System
Practice Questions
Cerebrovascular Diseases
Practice Questions
Trauma to the Central Nervous System
Practice Questions
Infections of the Nervous System
Practice Questions
Demyelinating Diseases
Practice Questions
Neurodegenerative Diseases
Practice Questions
CNS Tumors
Practice Questions
Peripheral Nerve Disorders
Practice Questions
Neuromuscular Junction Diseases
Practice Questions
Congenital and Developmental Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app