
Neuropathology — MCQs

On this page
Which of the following cell types does not participate in repair after brain infarction?
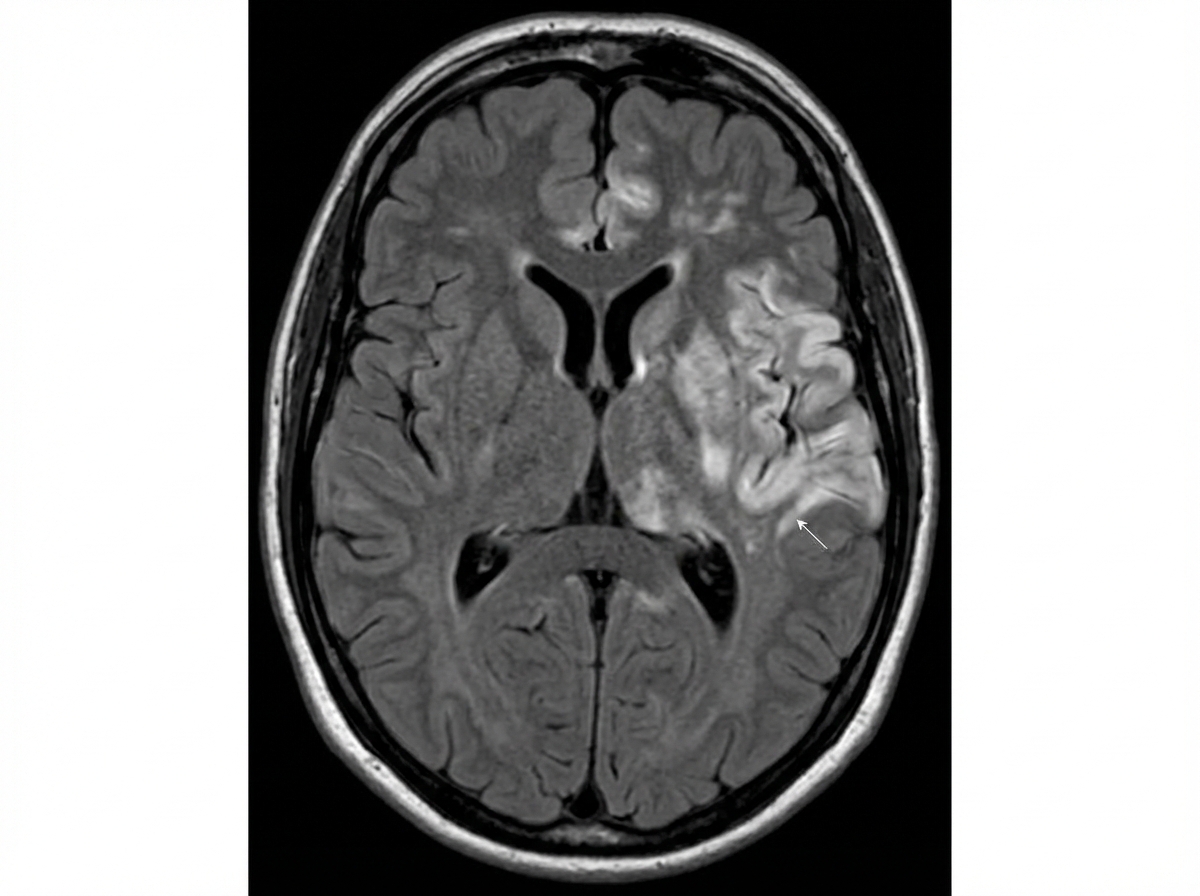
A 50-year-old man presented with progressive confusion, high fever, and somnolence. On examination, he was confused and hallucinating. After admission, he developed a tonic-clonic seizure. There were no focal neurological deficits. NCCT head showed no acute bleeding or elevated ICP. CSF analysis showed elevated protein and lymphocytic leukocytosis. MRI brain was performed. Which of the following histopathological patterns is most commonly associated with this condition?
Medulloblastoma exclusively occurs in which location?
What organ is typically associated with the presence of a D u r c k granuloma?
An 18-year-old man suffers massive trauma in a motorcycle accident. A CT scan shows multiple intracerebral hemorrhages. The patient expires after 6 months in a coma. At autopsy, there are cystic cavities within the frontal and temporal lobes, corresponding to the areas of prior hemorrhage. These cavities were formed in large measure due to the phagocytic activity of which of the following cell types?
What is the most common slowly growing vascular tumor of the spinal cord, cerebellum, and brain?
Which is the most common childhood central nervous system (CNS) tumor to metastasize outside the brain?
Common posterior cranial fossa tumors include all of the following except?
Histologic sections from a mass originating from the meninges would most likely reveal which of the following findings?
Dürck's granuloma is seen in which organ?
Practice by Chapter
Cellular Pathology of the Nervous System
Practice Questions
Cerebrovascular Diseases
Practice Questions
Trauma to the Central Nervous System
Practice Questions
Infections of the Nervous System
Practice Questions
Demyelinating Diseases
Practice Questions
Neurodegenerative Diseases
Practice Questions
CNS Tumors
Practice Questions
Peripheral Nerve Disorders
Practice Questions
Neuromuscular Junction Diseases
Practice Questions
Congenital and Developmental Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app