
Cardiology — MCQs

On this page
In Marfan's syndrome, aortic aneurysm occurs most commonly in which part of the aorta?
Which of the following is true about symptomatic mitral valve prolapse?
The echocardiogram of a patient presenting with exertional dyspnea, bilateral pedal edema, and elevated jugular venous pressure is shown. What is the most likely diagnosis?
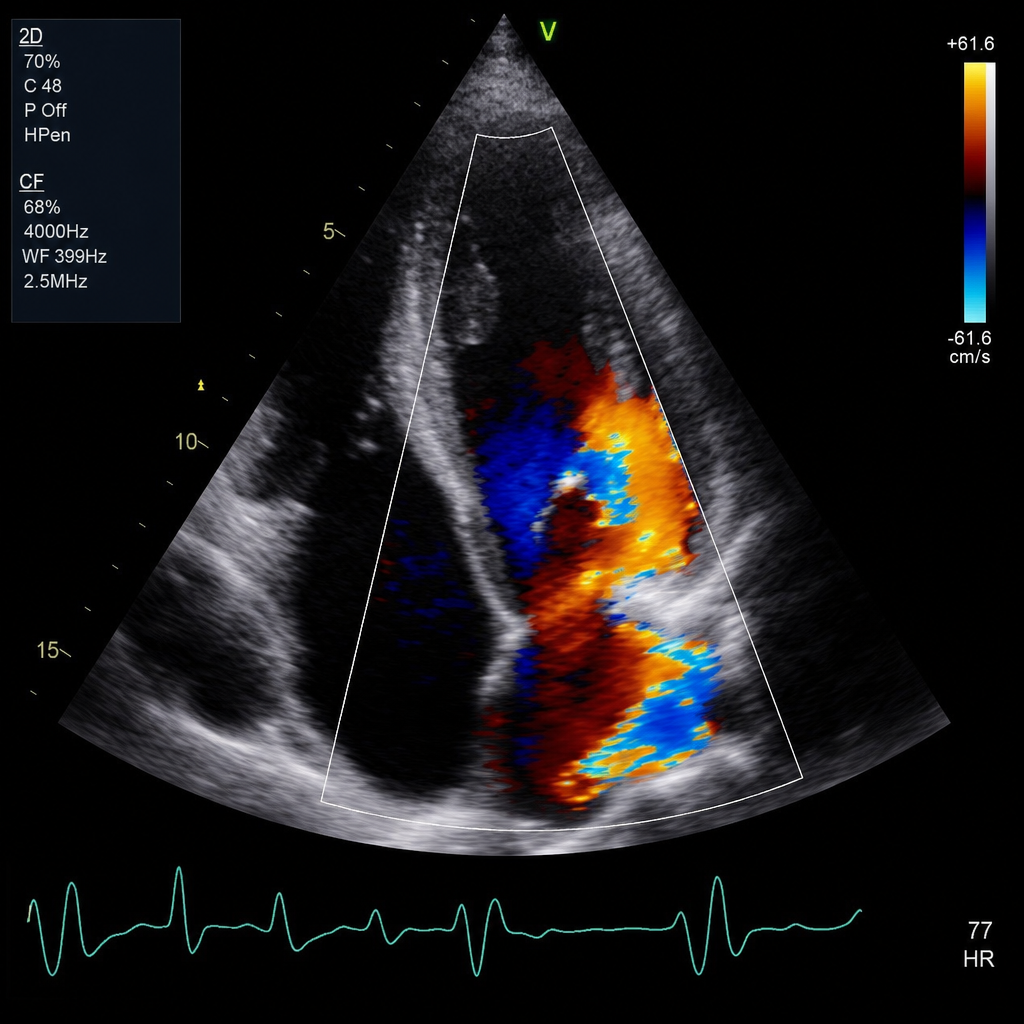
All of the following conditions produce restrictive cardiomyopathy except?
Calcification of the aortic valve is seen in which condition?
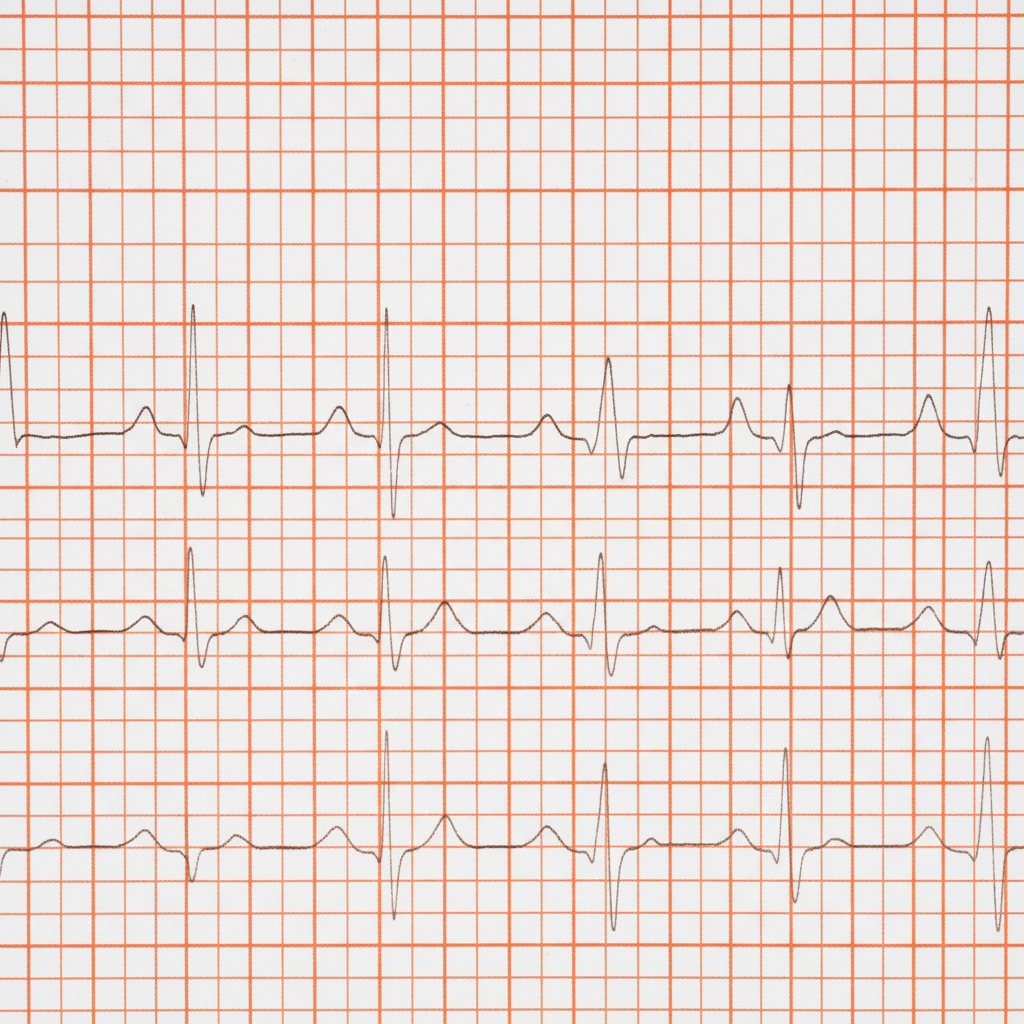
A 65-year-old man with diabetes, on an oral hypoglycemic, presents to the ER with a spontaneous right shoulder injury. His heart rate was noted to be irregular and the following ECG was obtained. What is the best immediate therapy?
A 35-year-old lady has Idiopathic pulmonary artery hypertension. Which findings best describe this patient?
A smoker presents with a four-year history of blanching of the fingers on exposure to cold. What is the most likely diagnosis?
What is the most common cause of morbidity and mortality late in the course of mitral stenosis?
All of the following statements about Brugada syndrome are true, EXCEPT:
Practice by Chapter
Coronary Artery Disease and Angina
Practice Questions
Acute Coronary Syndromes
Practice Questions
Heart Failure
Practice Questions
Cardiac Arrhythmias
Practice Questions
Valvular Heart Diseases
Practice Questions
Cardiomyopathies
Practice Questions
Pericardial Diseases
Practice Questions
Congenital Heart Disease in Adults
Practice Questions
Hypertension and Hypertensive Emergencies
Practice Questions
Pulmonary Hypertension
Practice Questions
Non-invasive Cardiac Diagnostics
Practice Questions
Preventive Cardiology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app