
Carbohydrate Metabolism — MCQs

On this page
Which of the following acts as a primer and accepts glucose residues during glycogenesis?
A 3-year-old boy has reduced red blood cell (RBC) numbers with minimal signs of anemia. Analysis of labeled RBCs shows a greatly reduced ATP yield compared to individuals without anemia. Which of the following would be expected to increase in the RBCs of this child?
Which of the following is true regarding the pathway shown in the figure?
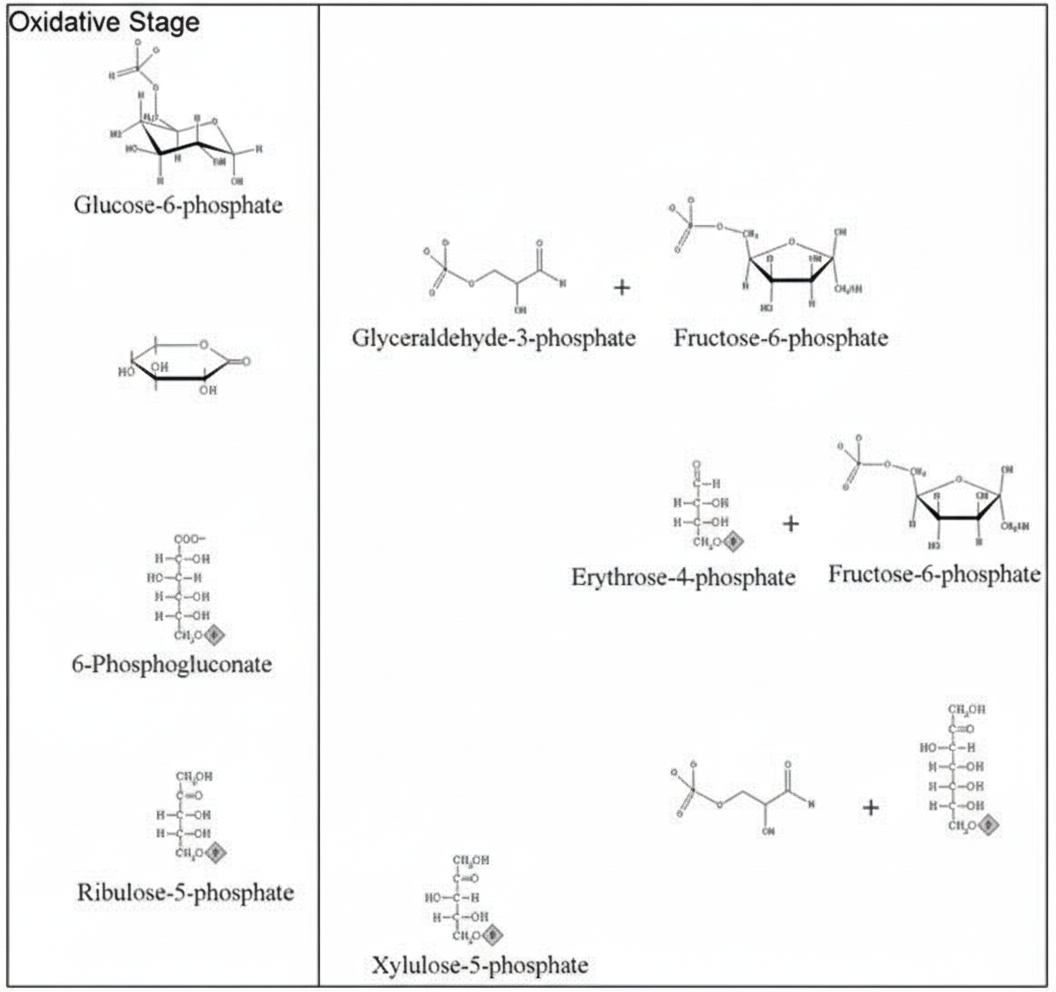
How many ATPs are directly produced by the Hexose Monophosphate (HMP) shunt pathway?
Which of the following represents the primary function of the pentose phosphate pathway in erythrocytes?
What is the primary product of the pentose phosphate pathway besides pentoses?
In anaerobic glycolysis, what is the net gain of ATP?
Which of the following intermediates of the TCA cycle is depleted in Type-I Diabetes mellitus to suppress the TCA cycle?
What is the carbohydrate component of blood group substances?
In one cycle of glycolysis under aerobic conditions, how many molecules of ATP and NADPH are formed?
Practice by Chapter
Carbohydrate Chemistry and Classification
Practice Questions
Glycolysis: Reactions and Regulation
Practice Questions
Gluconeogenesis: Reactions and Regulation
Practice Questions
Glycogen Metabolism: Synthesis and Breakdown
Practice Questions
Glycogen Storage Diseases
Practice Questions
Pentose Phosphate Pathway
Practice Questions
Metabolism of Fructose and Galactose
Practice Questions
Disorders of Fructose and Galactose Metabolism
Practice Questions
Blood Glucose Regulation
Practice Questions
Diabetes Mellitus: Biochemical Aspects
Practice Questions
Glycosylation and Glycoproteins
Practice Questions
Lactose Intolerance and Galactosemia
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app