
Carbohydrate Metabolism — MCQs

On this page
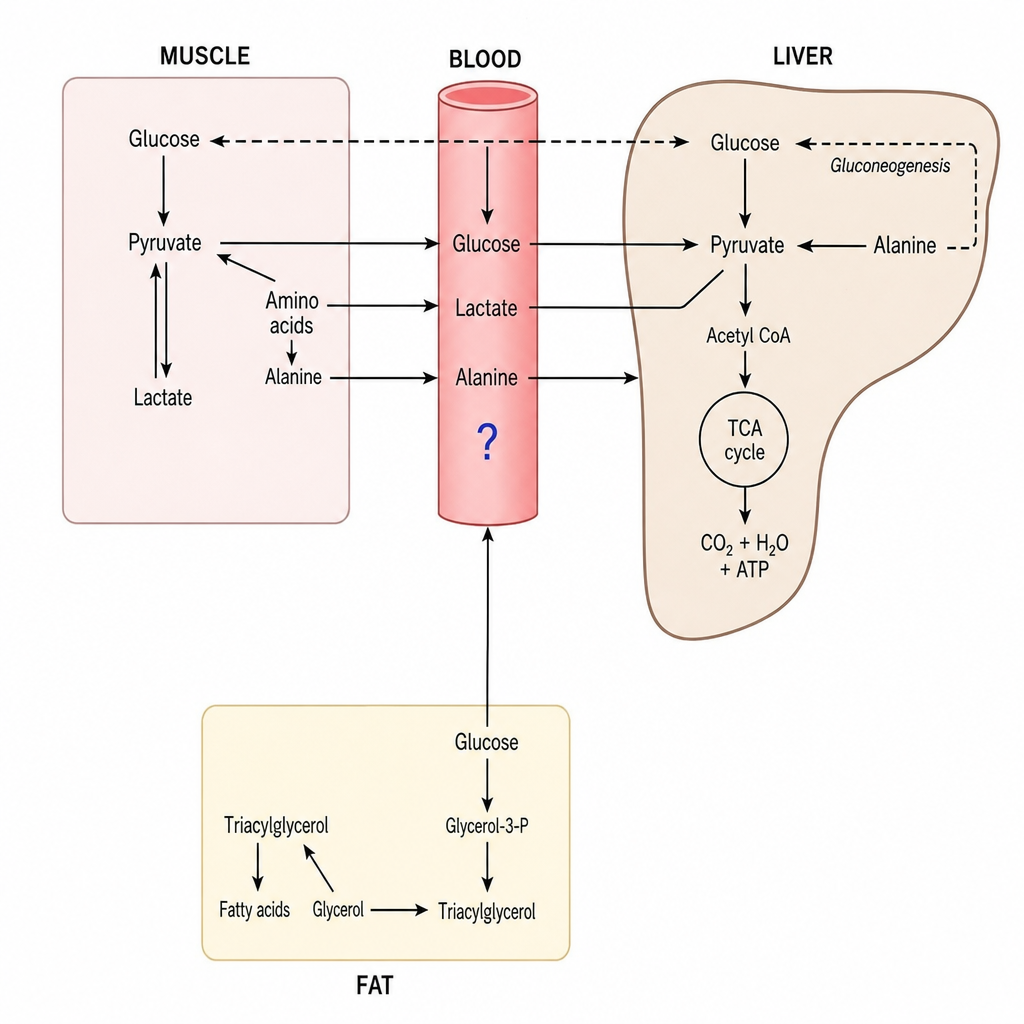
The molecule marked by a blue question mark can be all of the following EXCEPT?
A 3-month-old infant presents with hepatosplenomegaly and failure to thrive. A liver biopsy reveals glycogen with an abnormal, amylopectin-like structure with long outer chains and missing branches. Which of the following enzymes would most likely be deficient?
Phosphorylase b is maintained in an inactivated state by?
A boy presents with vomiting, a bloated abdomen, and abdominal pain. He attended an ice-cream eating competition last night and has a past history of similar episodes following the ingestion of milk and milk products. What is the likely cause?
Which of the following statements is true regarding GLUT-5?
Which metabolic pathway provides the reducing agent primarily used in lipogenesis?
In a well-fed state, what is the major fate of glucose-6-phosphate in tissues?
Glucose can be synthesized from which of the following substrates?
The branching enzyme is involved in which process?
Which of the following enzymes does not participate in the TCA cycle?
Practice by Chapter
Carbohydrate Chemistry and Classification
Practice Questions
Glycolysis: Reactions and Regulation
Practice Questions
Gluconeogenesis: Reactions and Regulation
Practice Questions
Glycogen Metabolism: Synthesis and Breakdown
Practice Questions
Glycogen Storage Diseases
Practice Questions
Pentose Phosphate Pathway
Practice Questions
Metabolism of Fructose and Galactose
Practice Questions
Disorders of Fructose and Galactose Metabolism
Practice Questions
Blood Glucose Regulation
Practice Questions
Diabetes Mellitus: Biochemical Aspects
Practice Questions
Glycosylation and Glycoproteins
Practice Questions
Lactose Intolerance and Galactosemia
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app