
Amino Acid Metabolism — MCQs

On this page
A patient reports a change in colour of urine on air exposure. All are true about the condition shown below except:
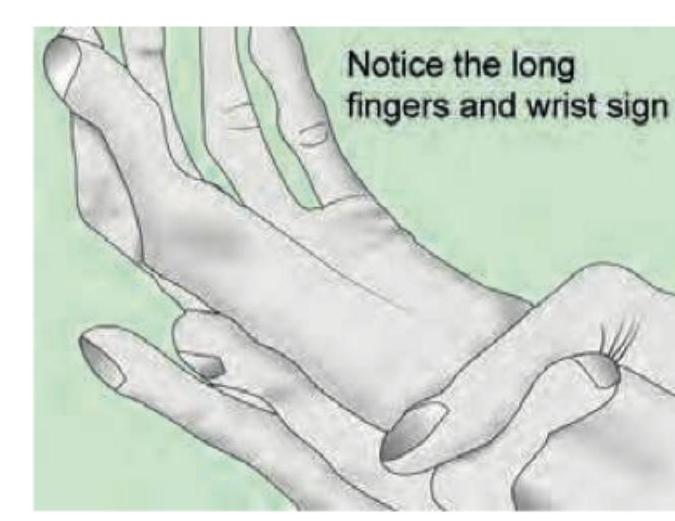
The image shows a tall patient with positive urine nitroprusside test diagnostic of Homocystinuria. Which of the following is true regarding Homocystinuria compared to Marfan's syndrome?
Of the following amino acids required by the human body, which one falls under the category of 'essential' amino acid?
In a patient with maple syrup urine disease, all of the following amino acids should be restricted in diet except?
Which of the following options is false in a patient with advanced liver disease?
Ammonia formed in the brain is converted into
Atherosclerosis is associated with:
Melanin is derived from which amino acid?
All are involved in non-toxic transport of ammonia except:
Glutathione does all of the following except?
Practice by Chapter
Protein Digestion and Absorption
Practice Questions
Transamination and Deamination
Practice Questions
Urea Cycle
Practice Questions
Disorders of Urea Cycle
Practice Questions
Metabolism of Individual Amino Acids
Practice Questions
Inborn Errors of Amino Acid Metabolism
Practice Questions
Phenylketonuria and Alkaptonuria
Practice Questions
Homocystinuria and Methionine Metabolism
Practice Questions
Synthesis of Biologically Important Compounds from Amino Acids
Practice Questions
Nitrogen Balance
Practice Questions
Ammonia Metabolism and Toxicity
Practice Questions
One-Carbon Transfer Reactions
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app