
Amino Acid Metabolism — MCQs

On this page
Nitric oxide is synthesized from which of the following amino acids?
A patient has elevated phenylalanine levels (40 mg/dL), but phenylalanine hydroxylase enzyme levels are normal. Which cofactor deficiency is most likely responsible for this condition?
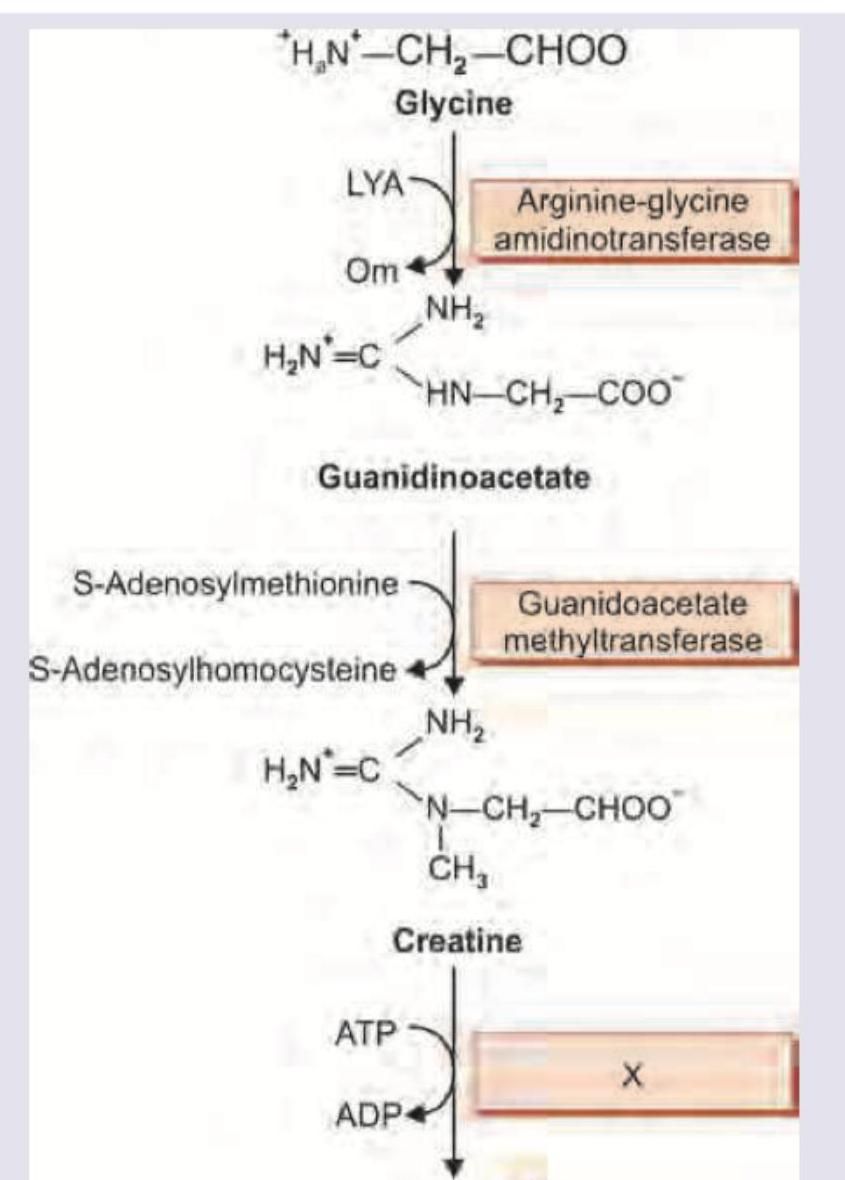
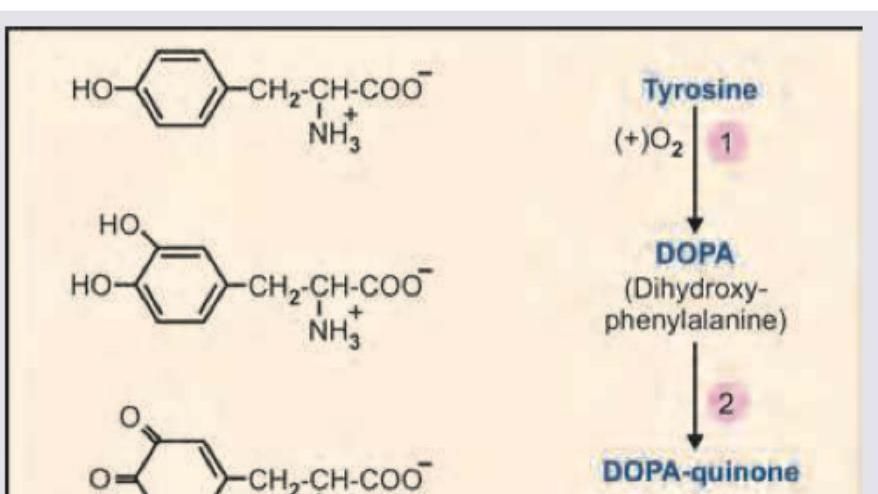
The image shows a biochemical pathway. Name the enzyme marked as X.
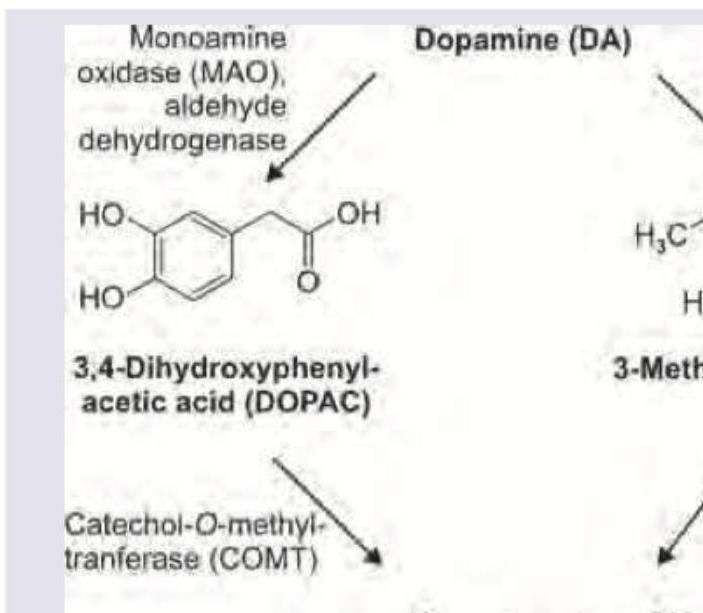
The image shows dopamine metabolism. Which is the product named as $X$ ?
A patient presents with ochronosis. Which of the following substance accumulates in this condition?
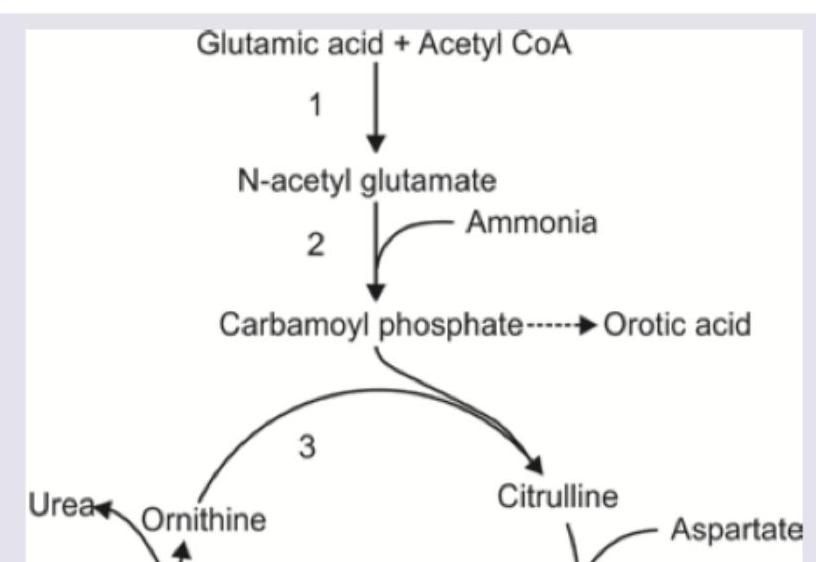
An infant presents with failure to thrive and recurrent vomiting episodes. Sepsis screen was negative, blood urea is elevated and urine shows high orotic acid level. Which of the following enzyme is absent?
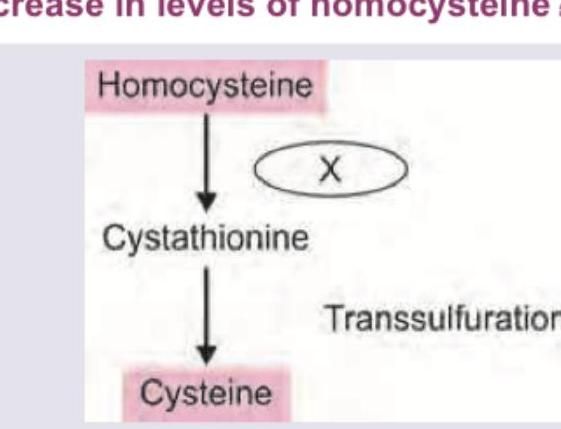
Deficiency of which of the following will lead to increase in levels of homocysteine?
Identify the amino acid marked as X from which all the products highlighted below are derived.
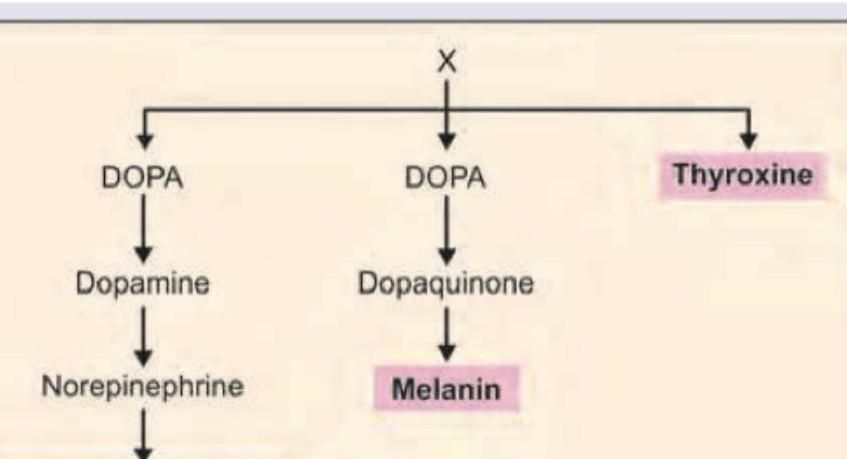
All are correct about the amino acid marked as X, EXCEPT:
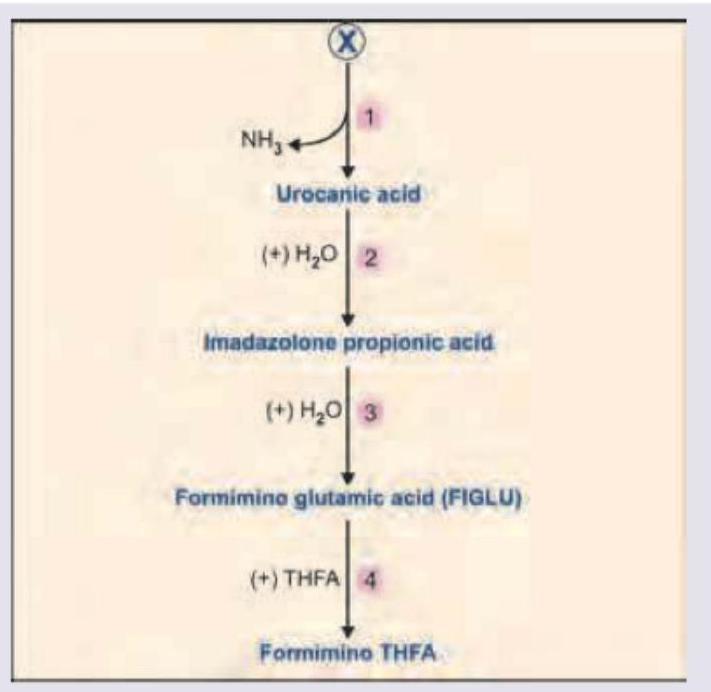
Name the product marked as X in the image shown below:
Practice by Chapter
Protein Digestion and Absorption
Practice Questions
Transamination and Deamination
Practice Questions
Urea Cycle
Practice Questions
Disorders of Urea Cycle
Practice Questions
Metabolism of Individual Amino Acids
Practice Questions
Inborn Errors of Amino Acid Metabolism
Practice Questions
Phenylketonuria and Alkaptonuria
Practice Questions
Homocystinuria and Methionine Metabolism
Practice Questions
Synthesis of Biologically Important Compounds from Amino Acids
Practice Questions
Nitrogen Balance
Practice Questions
Ammonia Metabolism and Toxicity
Practice Questions
One-Carbon Transfer Reactions
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app