
Amino Acid Metabolism — MCQs

On this page
OTC deficiency causes which of the following conditions?
Calcification of intervertebral discs is characteristic of which condition?
A patient with Gyrate Atrophy due to defective ornithine aminotransferase would be benefited by which dietary modification?
All of the following are ketogenic and glucogenic amino acids except?
Deficiency of arginosuccinate synthase causes which of the following disorders?
All of the following are characteristics of phenylalanine hydroxylase, EXCEPT?
"Marfan-like syndrome" is associated with which of the following conditions?
Which enzyme of the urea cycle is deficient in the brain?
What is the most direct precursor of taurine?
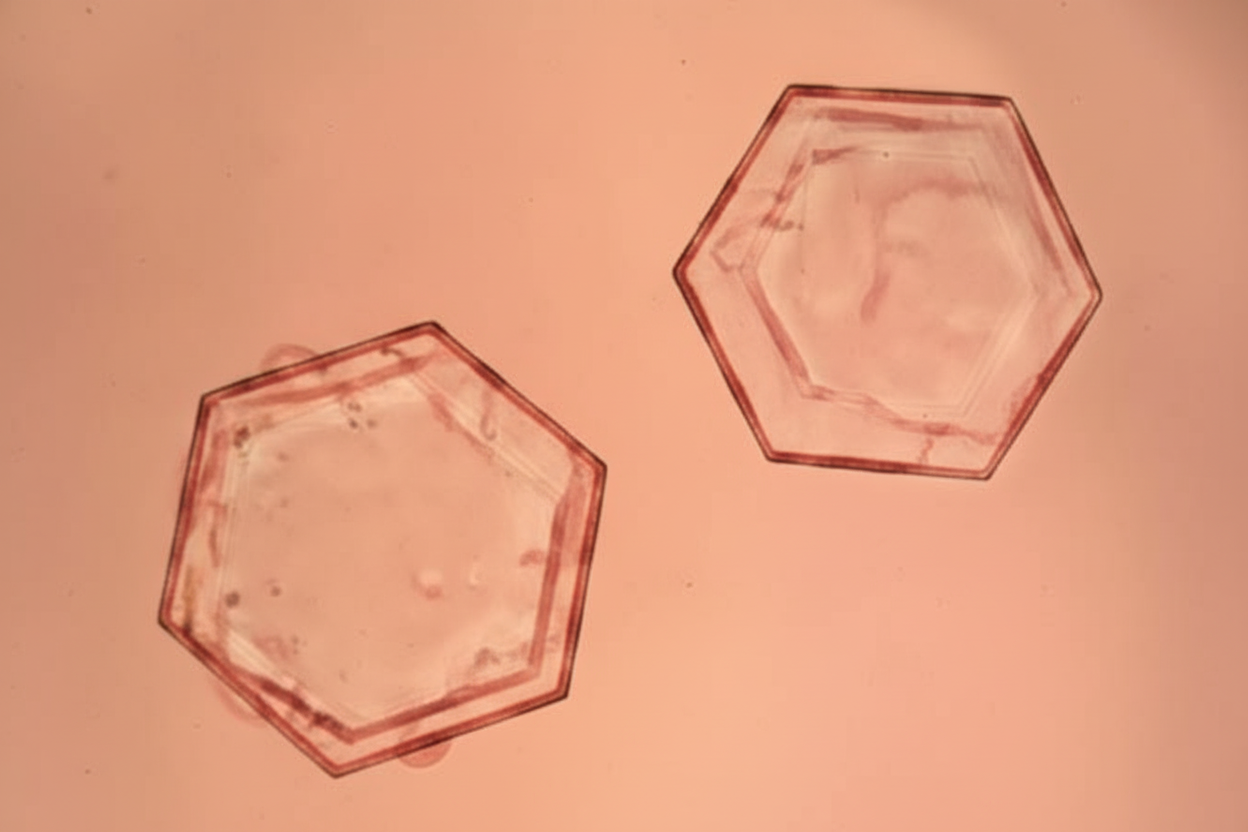
A patient presents with recurrent kidney stones. Microscopic examination of the urine specimen is shown below. Which of the following is not seen in the urine of this patient?
Practice by Chapter
Protein Digestion and Absorption
Practice Questions
Transamination and Deamination
Practice Questions
Urea Cycle
Practice Questions
Disorders of Urea Cycle
Practice Questions
Metabolism of Individual Amino Acids
Practice Questions
Inborn Errors of Amino Acid Metabolism
Practice Questions
Phenylketonuria and Alkaptonuria
Practice Questions
Homocystinuria and Methionine Metabolism
Practice Questions
Synthesis of Biologically Important Compounds from Amino Acids
Practice Questions
Nitrogen Balance
Practice Questions
Ammonia Metabolism and Toxicity
Practice Questions
One-Carbon Transfer Reactions
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app