
Systemic Pathology — MCQs

On this page
A 21-year-old male presents to your office with hematuria 3 days after the onset of a productive cough and fever. Following renal biopsy, immunofluorescence shows granular IgA deposits in the glomerular mesangium. Which of the following do you suspect in this patient?
A 42-year-old woman is brought to the emergency department because of two episodes of hemoptysis over the past 24 hours. The patient has a 6-month history of severe sinusitis and bloody nasal discharge. Her vital parameters are as follows: blood pressure, 155/75 mm Hg; pulse, 75/min; respiratory rate, 14/min; and temperature, 37.9°C (100.2°F). Examination reveals red conjunctiva, and an ulcer on the nasal septum. Pulmonary auscultation indicates diffuse rhonchi. Cardiac and abdominal examinations reveal no abnormalities. Laboratory studies show: Urine Blood 3+ Protein 2+ RBC 10-15/hpf with dysmorphic features RBC cast numerous Based on these findings, this patient is most likely to carry which of the following antibodies?
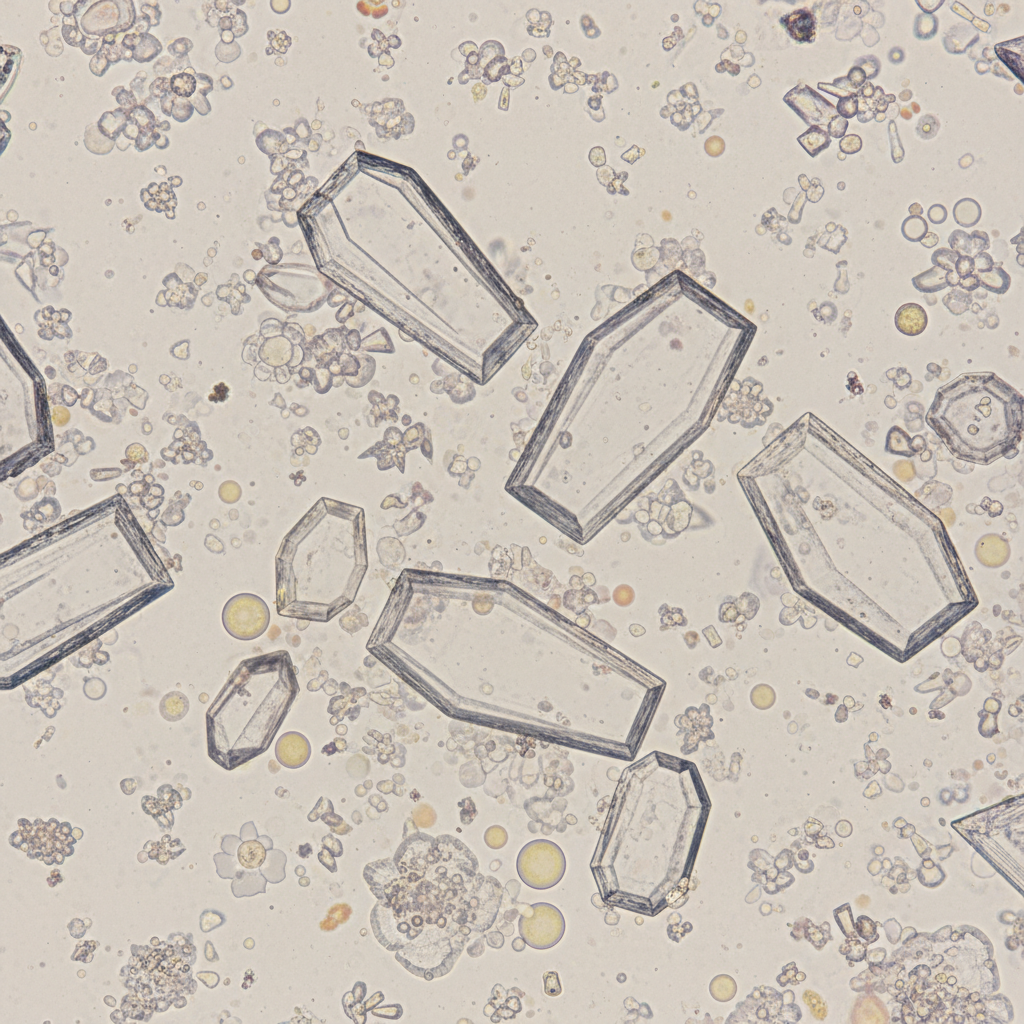
A 32-year-old woman comes to the physician because of a 1-week history of left flank pain and dysuria. She has had 2 episodes of urinary tract infection over the past 2 years. Her temperature is 37°C (98.6°F) and pulse is 82/min. An ultrasound of the kidneys shows left-sided hydronephrosis and echogenic foci with acoustic shadowing. A photomicrograph of the urine is shown. The crystals observed are most likely composed of which of the following?
A 10-year-old boy comes to the physician because of a 4-month history of intermittent red urine. During the past 2 years, he has had recurrent episodes of swelling of his face and feet. Five years ago, he was diagnosed with mild bilateral sensorineural hearing loss. His uncle died of kidney disease in his twenties. His blood pressure is 145/85 mm Hg. Laboratory studies show a hemoglobin concentration of 12.5 g/dL, urea nitrogen concentration of 40 mg/dL, and creatinine concentration of 2.4 mg/dL. Urinalysis shows 5–7 RBC/hpf. Which of the following is the most likely underlying cause of this patient's symptoms?
A 45-year-old male immigrant with rheumatoid arthritis comes to the physician because of severe pain and swelling in both his knees. He also reports an unintentional weight loss of around 10 kg over 3 months and episodic abdominal pain, varying in intensity and location. He has been having loose stools with no blood, 2–3 times a day for 1 month. He denies fever, night sweats, cough, or shortness of breath. Current medications include methotrexate, naproxen, and folic acid. His weight is 68 kg (150 lbs), temperature is 37.4°C (99.3°F), pulse is 90/min, and blood pressure is 130/80 mm Hg. Examination shows pale conjunctivae, cheilitis, and hyperpigmentation of the skin around his neck. Generalized lymphadenopathy is present. Examination of the knee joints shows bilateral warmth, erythema, swelling, tenderness, and limited range of motion. A grade 2/6 early diastolic murmur is heard over the right second intercostal space and an S3 is heard. Abdominal examination shows no abnormalities. Laboratory studies show: Hemoglobin 9.1 g/dL Leukocyte count 3800/mm3 Platelet count 140,000/mm3 Mean corpuscular volume 67 μm3 Erythrocyte sedimentation rate 62 mm/h Serum Glucose 100 mg/dL Creatinine 0.7 mg/dL TIBC 500 mcg/dL Ferritin 10 mcg/dL Rheumatoid factor negative Anti -CCP negative An esophagogastroduodenoscopy is ordered. A biopsy specimen of the duodenum is likely to show which of the following?
A 25-year-old man of Mediterranean descent makes an appointment with his physician because his skin and sclera have become yellow. He complains of fatigue and fever that started at the same time icterus appeared. On examination, he is tachycardic and tachypneic. The oxygen (O2) saturation is < 90%. He has increased unconjugated bilirubin, hemoglobinemia, and an increased number of reticulocytes in the peripheral blood. What is the most likely diagnosis?
A 27-year-old woman presents to the emergency department complaining of a left-sided headache and right-sided blurry vision. She states that 2 weeks ago she developed dark urine and abdominal pain. She thought it was a urinary tract infection so she took trimethoprim-sulfamethoxazole that she had left over. She planned on going to her primary care physician today but then she developed headache and blurry vision so she came to the emergency department. The patient states she is otherwise healthy. Her family history is significant for a brother with sickle cell trait. On physical examination, there is mild abdominal tenderness, and the liver edge is felt 4 cm below the right costal margin. Labs are drawn as below: Hemoglobin: 7.0 g/dL Platelets: 149,000/mm^3 Reticulocyte count: 5.4% Lactate dehydrogenase: 3128 U/L Total bilirubin: 2.1 mg/dL Indirect bilirubin: 1.4 mg/dL Aspartate aminotransferase: 78 U/L Alanine aminotransferase: 64 U/L A peripheral smear shows polychromasia. A Doppler ultrasound of the liver shows decreased flow in the right hepatic vein. Magnetic resonance imaging of the brain is pending. Which of the following tests, if performed, would most likely identify the patient’s diagnosis?
A 3080-g (6-lb 13-oz) male newborn is delivered at term to a 27-year-old woman, gravida 2, para 1. Pregnancy was uncomplicated. He appears pale. His temperature is 36.8°C (98.2°F), pulse is 167/min, and respirations are 56/min. Examination shows jaundice of the skin and conjunctivae. The liver is palpated 2–3 cm below the right costal margin, and the spleen is palpated 1–2 cm below the left costal margin. The lungs are clear to auscultation. No murmurs are heard. His hemoglobin concentration is 10.6 g/dL and mean corpuscular volume is 73 μm3. Hemoglobin DNA testing shows 3 missing alleles. Which of the following laboratory findings is most likely present in this patient?
A 61-year-old man presents to the emergency department because he has developed blisters at multiple locations on his body. He says that the blisters appeared several days ago after a day of hiking in the mountains with his colleagues. When asked about potential triggering events, he says that he recently had an infection and was treated with antibiotics but he cannot recall the name of the drug that he took. In addition, he accidentally confused his medication with one of his wife's blood thinner pills several days before the blisters appeared. On examination, the blisters are flesh-colored, raised, and widespread on his skin but do not involve his mucosal surfaces. The blisters are tense to palpation and do not separate with rubbing. Pathology of the vesicles show that they continue under the level of the epidermis. Which of the following is the most likely cause of this patient's blistering?
A 61-year-old man comes to the physician because of progressively worsening swelling of his ankles. He says he has felt exhausted lately. Over the past 3 months, he has gained 5 kg. He has smoked one pack of cigarettes daily for 30 years. His pulse is 75/min and his blood pressure is 140/90 mmHg. Examination shows 2+ pitting edema in the lower extremities. Neurologic exam shows diminished two-point discrimination in the fingers and toes. A urine sample is noted to be foamy. Laboratory studies show a hemoglobin A1c of 7.9% and creatinine of 1.9 mg/dL. A biopsy specimen of the kidney is most likely to show which of the following?
Practice by Chapter
Liver pathology (hepatitis, cirrhosis)
Practice Questions
Gallbladder and biliary tract disorders
Practice Questions
Pancreatic diseases
Practice Questions
Kidney diseases
Practice Questions
Male reproductive pathology
Practice Questions
Female reproductive pathology
Practice Questions
Breast pathology
Practice Questions
Endocrine pathology
Practice Questions
Bone and joint pathology
Practice Questions
Skeletal muscle diseases
Practice Questions
Peripheral nerve disorders
Practice Questions
Soft tissue tumors
Practice Questions
Head and neck pathology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app