
Systemic Pathology — MCQs

On this page
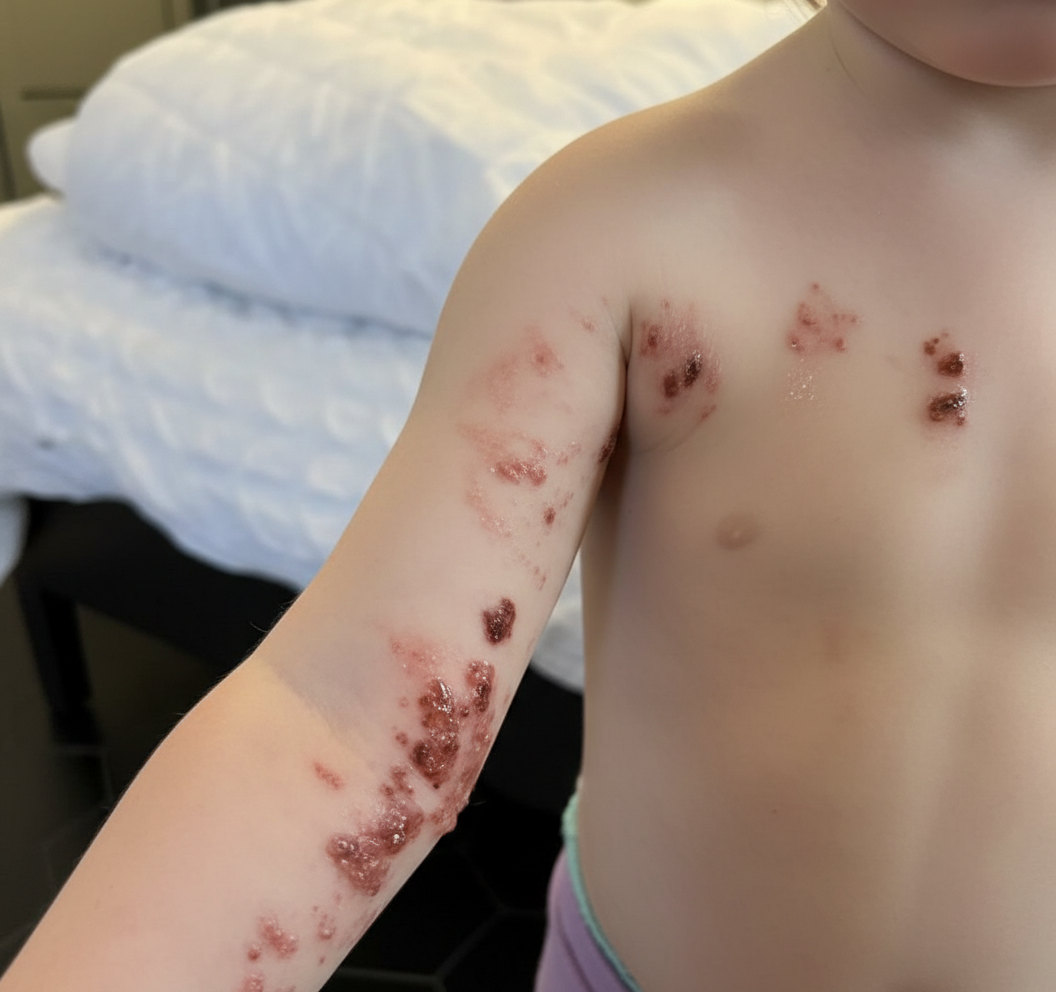
A 5-year-old boy is brought to the physician because of a painful, burning rash on his left arm for 3 days. Three years ago, he was diagnosed with heart failure due to congenital heart disease and received an allogeneic heart transplantation. He takes cyclosporine to prevent chronic transplant rejection. He has not received any routine childhood vaccinations. A photograph of the rash is shown. Microscopic examination of a skin biopsy specimen is most likely to show which of the following findings?
A 21-year-old man comes to the physician because of a 3-day history of yellowing of his eyes. He has also noticed a decrease in his exercise capacity and gets quickly exhausted after minor physical activity. Examination shows scleral icterus and pale mucous membranes. He has splenomegaly. His hemoglobin concentration is 7.9 g/dL, leukocyte is count 8500/mm3, and platelet count is 187,000/mm3. Direct antiglobulin and heterophile antibody tests are positive. Which of the following additional laboratory findings are most likely present in this patient?
A pathologist performed an autopsy on an 18-month-old infant boy who died of pneumonia. Clinical notes revealed the infant had repeated respiratory infections that started after he was weaned off of breast-milk. Laboratory investigation revealed hypogammaglobulinemia and an absence of B-cells. T-cell levels were normal. Histological evaluation of an axillary lymph node revealed an absence of germinal centers. Which of the following is the mode of inheritance of the disorder that afflicted this infant?
A 12-year-old boy comes to the physician for the evaluation of intermittent blood-tinged urine for several months. Four months ago, he had an episode of fever and sore throat that resolved without treatment after 5 days. During the past 2 years, he has also had recurrent episodes of swelling of his face and feet. 5 years ago, he was diagnosed with mild bilateral sensorineural hearing loss. His brother died of a progressive kidney disease at the age of 23. The patient appears pale. His temperature is 37°C (98.6°F), pulse is 70/min, and blood pressure is 145/85 mm Hg. Slit lamp examination shows a conical protrusion of both lenses. Laboratory studies show a hemoglobin concentration of 11 g/dL, urea nitrogen concentration of 40 mg/dL, and creatinine concentration of 2.4 mg/dL. Urinalysis shows: Blood 2+ Protein 1+ RBC 5–7/hpf RBC casts rare Which of the following is the most likely underlying cause of this patient's symptoms?
A 3-month-old is referred to a pediatric immunologist by his pediatrician for further workup of recurrent sinopulmonary infections which have not abated despite adequate treatment. During the workup flow cytometry demonstrates a decrease in normal CD40L cells. Based on these findings, the immunologist decides to pursue a further workup and obtains immunoglobulin levels. Which of the following immunoglobulin profiles is most likely to be observed in this patient?
A 31-year-old man presents to the office with complaints of multiple episodes of blood in his urine as well as coughing of blood for the past 3 days. He also reports a decrease in urinary frequency, and denies pain with urination. No previous similar symptoms or significant past medical history is noted. There is no history of bleeding disorders in his family. His vitals include a blood pressure of 142/88 mm Hg, a pulse of 87/min, a temperature of 36.8°C (98.2°F), and a respiratory rate of 11/min. On physical examination, chest auscultation reveals normal vesicular breath sounds. Abdominal exam is normal. The laboratory results are as follows: Complete blood count Hemoglobin 12 g/dL RBC 4.9 x 106 cells/µL Hematocrit 48% Total leukocyte count 6,800 cells/µL Neutrophils 70% Lymphocyte 25% Monocytes 4% Eosinophil 1% Basophils 0% Platelets 200,000 cells/µL Urine examination pH 6.2 Color dark brown RBC 18–20/HPF WBC 3–4/HPF Protein 1+ Cast RBC casts Glucose absent Crystal none Ketone absent Nitrite absent 24 hours urine protein excretion 1.3 g A renal biopsy under light microscopy shows a crescent formation composed of fibrin and macrophages. Which of the following best describes the indirect immunofluorescence finding in this condition?
A 9-year-old boy is brought to the emergency room by his mother. She is concerned because her son’s face has been swollen over the past 2 days. Upon further questioning, the boy reports having darker urine without dysuria. The boy was seen by his pediatrician 10 days prior to presentation with a crusty yellow sore on his right upper lip that has since resolved. His medical history is notable for juvenile idiopathic arthritis. His temperature is 99°F (37.2°C), blood pressure is 140/90 mmHg, pulse is 95/min, and respirations are 18/min. On exam, he has mild periorbital edema. Serological findings are shown below: C2: Normal C3: Decreased C4: Normal CH50: Decreased Additional workup is pending. This patient most likely has a condition caused by which of the following?
A 38-year-old man presents to the outpatient clinic for an annual employee health checkup. He does not have any complaints at the moment except for skin changes, as seen in the following image. He denies any history of trauma. His medical history is insignificant. His family history is negative for any skin disorders or autoimmune disease. He is a non-smoker and does not drink alcohol. Which of the following is the most likely mechanism for this presentation?

A young Caucasian couple in their late twenties present for an infertility evaluation after trying to conceive over 2 years. On physical exam, the female appears healthy and states that she has regular menstrual cycles. The male partner is noted to have long extremities with wide hips, low muscle mass, gynecomastia, sparse facial or chest hair, and small, firm testes. Laboratory tests of the male partner reveal elevated serum LH and FSH and low testosterone levels. If cytogenetic tests were performed, which of the following would be seen in this male?
A 10-year-old boy is presented to the hospital for a kidney transplant. In the operating room, the surgeon connects an allograft kidney renal artery to the aorta, and after a few moments, the kidney becomes cyanotic, edematous, and dusky with mottling. Which of the following in the recipient’s serum is responsible for this rejection?
Practice by Chapter
Liver pathology (hepatitis, cirrhosis)
Practice Questions
Gallbladder and biliary tract disorders
Practice Questions
Pancreatic diseases
Practice Questions
Kidney diseases
Practice Questions
Male reproductive pathology
Practice Questions
Female reproductive pathology
Practice Questions
Breast pathology
Practice Questions
Endocrine pathology
Practice Questions
Bone and joint pathology
Practice Questions
Skeletal muscle diseases
Practice Questions
Peripheral nerve disorders
Practice Questions
Soft tissue tumors
Practice Questions
Head and neck pathology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app