
Systemic Pathology — MCQs

On this page
A 17-year-old boy is brought to the physician because of swelling of his face and legs for 5 days. He immigrated to the United States from Korea with his family 10 years ago. He has been healthy except for an episode of sore throat 2 weeks ago. His younger sister has type 1 diabetes mellitus. His temperature is 37°C (98.6°F), pulse is 90/min, and blood pressure is 145/87 mm Hg. Examination shows periorbital edema and 3+ pitting edema of the lower extremities. Laboratory studies show: Hemoglobin 13.9 g/dL Leukocyte count 8,100/mm3 Serum Glucose 78 mg/dL Albumin 2.4 g/dL Hepatitis B surface antigen positive Hepatitis B surface antibody negative Complement C4 decreased Urine Blood negative Protein 4+ Glucose negative Protein/creatinine ratio 8.1 (N ≤ 0.2) Further evaluation is most likely to show which of the following additional findings?
A 17-year-old boy is brought to the physician with complaints of an ataxic gait and hearing deficits for the past few days. His parents also reported a history of tonic gaze deviation on the right side and the spontaneous remission of a similar episode 6 months ago. His temperature is 37°C (98.6°F), pulse is 88/min, and respirations are 20/min. On physical examination, no abnormality is found, but evoked potential tests are abnormal. Magnetic resonance imaging of the head shows multiple lesions with high T2 signal intensity and one large white matter lesion showing contrast enhancement. His laboratory studies show: Hemoglobin 12.9 g/dL CSF leukocyte count 1000/μL CSF gamma globulin 15.4% (normal 7–14%) Erythrocyte sedimentation rate 16 mm/h Which of the following most likely explains the mechanism of this condition?
An investigator studying immune-mediated pulmonary damage performs an autopsy on a bilateral lung transplant recipient who died of hypercapnic respiratory failure. The patient underwent lung transplantation for idiopathic pulmonary fibrosis. Microscopic examination of the lung shows diffuse eosinophilic scarring of the terminal and respiratory bronchioles and near-complete luminal obliteration by polypoidal plugs of granulation tissue. Examination of the skin shows no abnormalities. The findings in this patient are most consistent with which of the following conditions?
A 45-year-old woman with β-thalassemia major comes to the physician with a 1-week history of fatigue. She receives approximately 8 blood transfusions per year; her last transfusion was 1 month ago. Examination shows conjunctival pallor. Her hemoglobin level is 6.5 mg/dL. Microscopic evaluation of a liver biopsy specimen in this patient would most likely show which of the following?
A 29-year-old woman presents to the office with the complaint of a tingling sensation over her face and distal parts of her lower limbs. Three weeks ago, she had an episode of bloody diarrhea and was successfully treated with erythromycin. She is a full-time radiology technician. Currently, she takes oral contraceptives and zopiclone (1 mg) at bedtime. Her blood pressure is 100/80 mm Hg, her heart rate is 91/min, her respiratory rate is 15/min, and her temperature is 36.7°C (98.0°F). Neurological examination reveals loss of all sensation over the face and in the distal part of her lower limbs. Strength in calf flexor and extensor muscles is diminished bilaterally (4/5 on all of the muscle groups). Deep tendon reflexes are 1+ in the knees and 1+ in the ankles. Plantar reflexes are flexor. What is the most probable mechanism of the pathological findings in this patient?
A 22-year-old man has had dyspnea and hemoptysis for the past week. He has no known sick contacts. There is no personal or family history of serious illness. He takes no medications. His temperature is 37°C (98.6°F), pulse is 82/min, respirations are 22/min, and blood pressure is 152/90 mm Hg. Examination shows inspiratory crackles at both lung bases. The remainder of the examination shows no abnormalities. His hemoglobin is 14.2 g/dL, leukocyte count is 10,300/mm3, and platelet count is 205,000/mm3. Urinalysis shows a proteinuria of 2+, 70 RBC/hpf, and 1–2 WBC/hpf. Chest x-ray shows pulmonary infiltrates. Further evaluation is most likely to show which of the following findings?
A 36-year-old man presents with massive hematemesis. Past medical history is significant for a gastric ulcer. He has a pulse of 115/min, respiratory rate of 20/min, temperature of 36°C (96.8°F), and blood pressure of 90/59 mm Hg. The patient receives a transfusion of 2 units of packed red blood cells. Around 5–10 minutes after the transfusion, he starts having chills, pain in the lumbar region, and oliguria. His vital signs change to pulse of 118/min, respiratory rate of 19/min, temperature of 38°C (100.4°F), and blood pressure of 60/40 mm Hg. Which of the following is the most likely cause of this patient’s condition?
A 59-year-old patient presented to his family physician 8 years ago with initial complaints of increasing generalized stiffness with trouble initiating movement and worsening micrographia. He was started on levodopa after further evaluation led to a suspected diagnosis of Parkinson's disease; however, this therapy ultimately failed to improve the patient's symptoms. Additionally, over the ensuing 8 years since his initial presentation, the patient also developed symptoms including worsening balance, orthostatic hypotension, urinary incontinence, and impotence. The patient's overall condition deteriorated ever since this initial diagnosis with increasing disability from his motor symptoms, and he recently passed away at the age of 67, 8 years after his first presentation to his physician. The family requests an autopsy. Which of the following would be expected on autopsy evaluation of this patient's brain tissue?
A 10-year-old boy presents to your office with puffy eyes. The patient's mother states that his eyes seem abnormally puffy and thinks he may have an eye infection. Additionally, he had a sore throat a week ago which resolved with over the counter medications. The mother also thought that his urine was darker than usual and is concerned that blood may be present. His temperature is 99.5°F (37.5°C), blood pressure is 107/62 mmHg, pulse is 100/min, respirations are 17/min, and oxygen saturation is 98% on room air. Physical exam is notable for bilateral periorbital edema. Cranial nerves are grossly intact bilaterally. Which of the following is the most likely finding on renal biopsy for this patient?
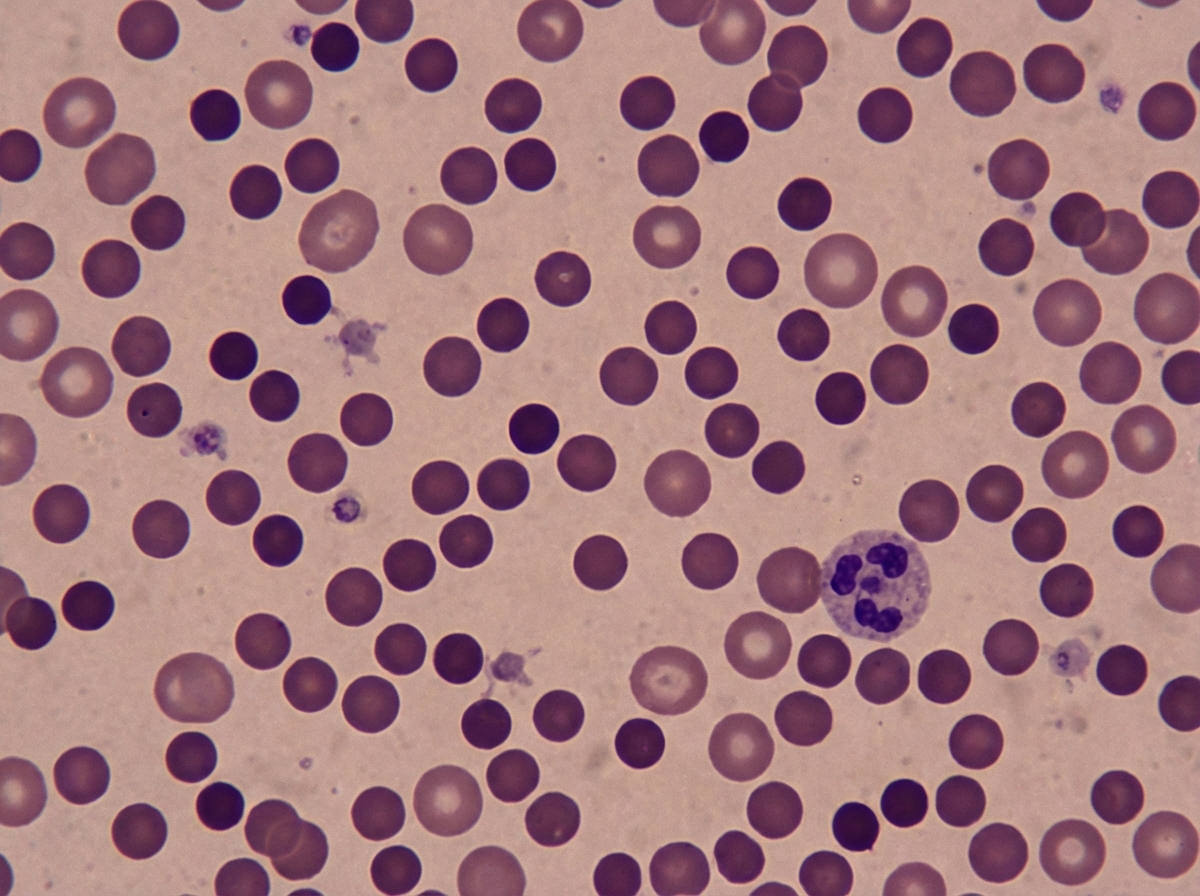
A 37-year-old man, otherwise healthy, has a routine CBC done prior to donating blood for the first time. The results are as follows: Hemoglobin 10.8 g/dL Mean corpuscular volume (MCV) 82 μm3 Mean corpuscular hemoglobin concentration (MCHC) 37% Reticulocyte count 3.2% White blood cell count 8,700/mm3 Platelet count 325,000/mm3 The patient is afebrile and his vital signs are within normal limits. On physical examination, his spleen is just palpable. A peripheral blood smear is shown in the exhibit (see image). A direct antiglobulin test (DAT) is negative. Which of the following best describes the etiology of this patient's most likely diagnosis?
Practice by Chapter
Liver pathology (hepatitis, cirrhosis)
Practice Questions
Gallbladder and biliary tract disorders
Practice Questions
Pancreatic diseases
Practice Questions
Kidney diseases
Practice Questions
Male reproductive pathology
Practice Questions
Female reproductive pathology
Practice Questions
Breast pathology
Practice Questions
Endocrine pathology
Practice Questions
Bone and joint pathology
Practice Questions
Skeletal muscle diseases
Practice Questions
Peripheral nerve disorders
Practice Questions
Soft tissue tumors
Practice Questions
Head and neck pathology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app