
Systemic Pathology — MCQs

On this page
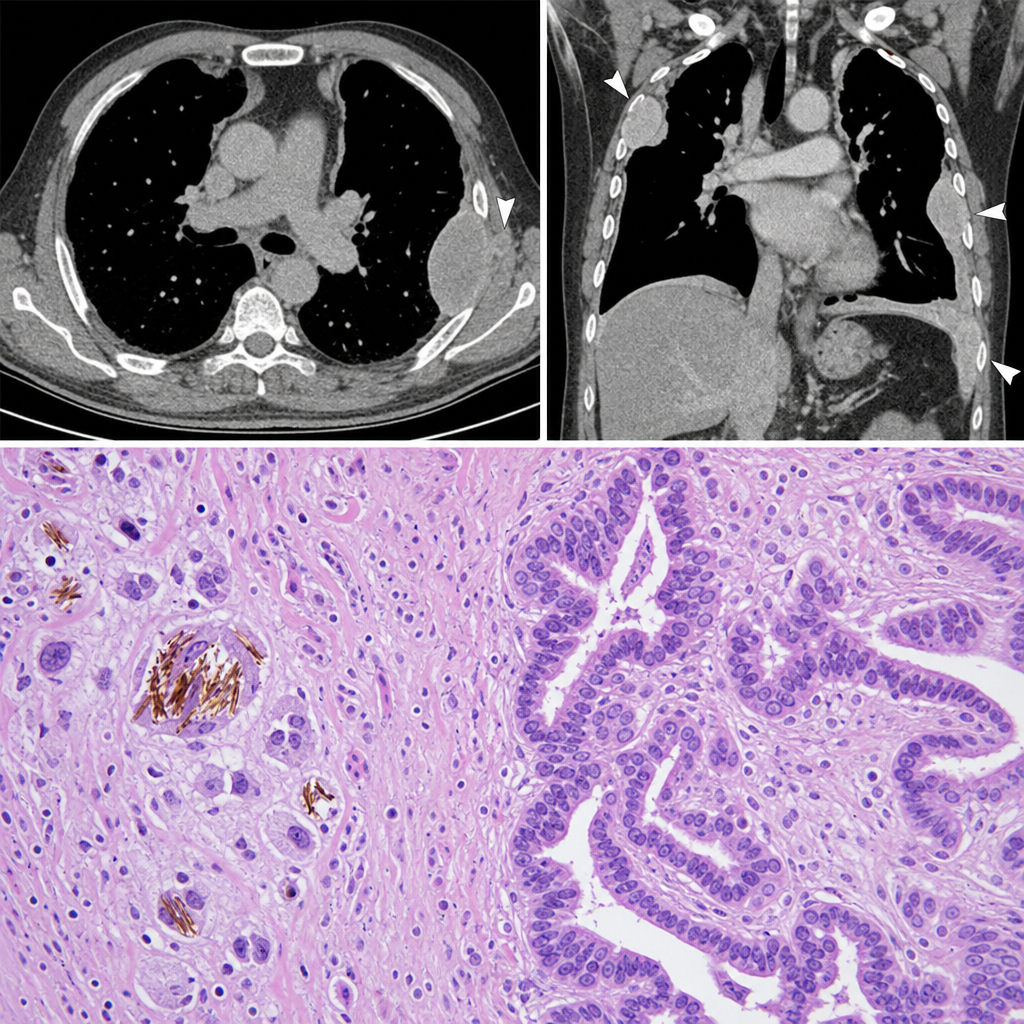
A 58-year-old man with a 40-pack-year smoking history presents with progressive dyspnea and a dry cough. Chest CT reveals bilateral pleural plaques and a right pleural mass. A pleural lesion biopsy is performed. The photomicrograph shows elongated, beaded golden-brown structures within macrophages, surrounded by a fibrous stroma with atypical mesothelial cells forming tubular and papillary patterns. Which of the following best describes the structures characteristic of asbestos exposure identified in this biopsy?
A 34-year-old woman presents with cervical lymphadenopathy, night sweats, and a mediastinal mass on chest X-ray. Excisional biopsy of a cervical lymph node is performed. The biopsy shows a mixed inflammatory background of lymphocytes, eosinophils, plasma cells, and neutrophils, within which large binucleated cells with prominent eosinophilic 'owl-eye' nucleoli are identified. Immunohistochemistry is performed. Which of the following immunophenotypic profiles is most consistent with the large binucleated cells seen in this biopsy?
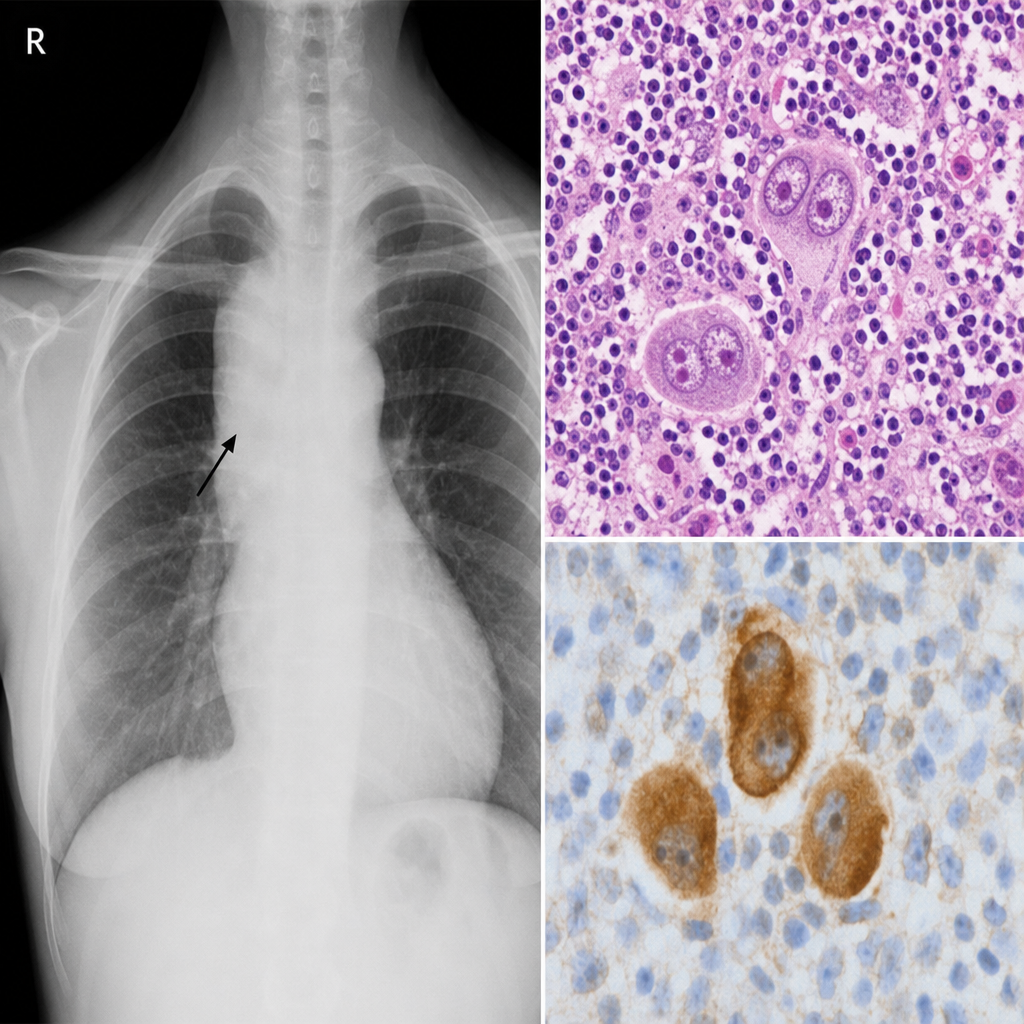
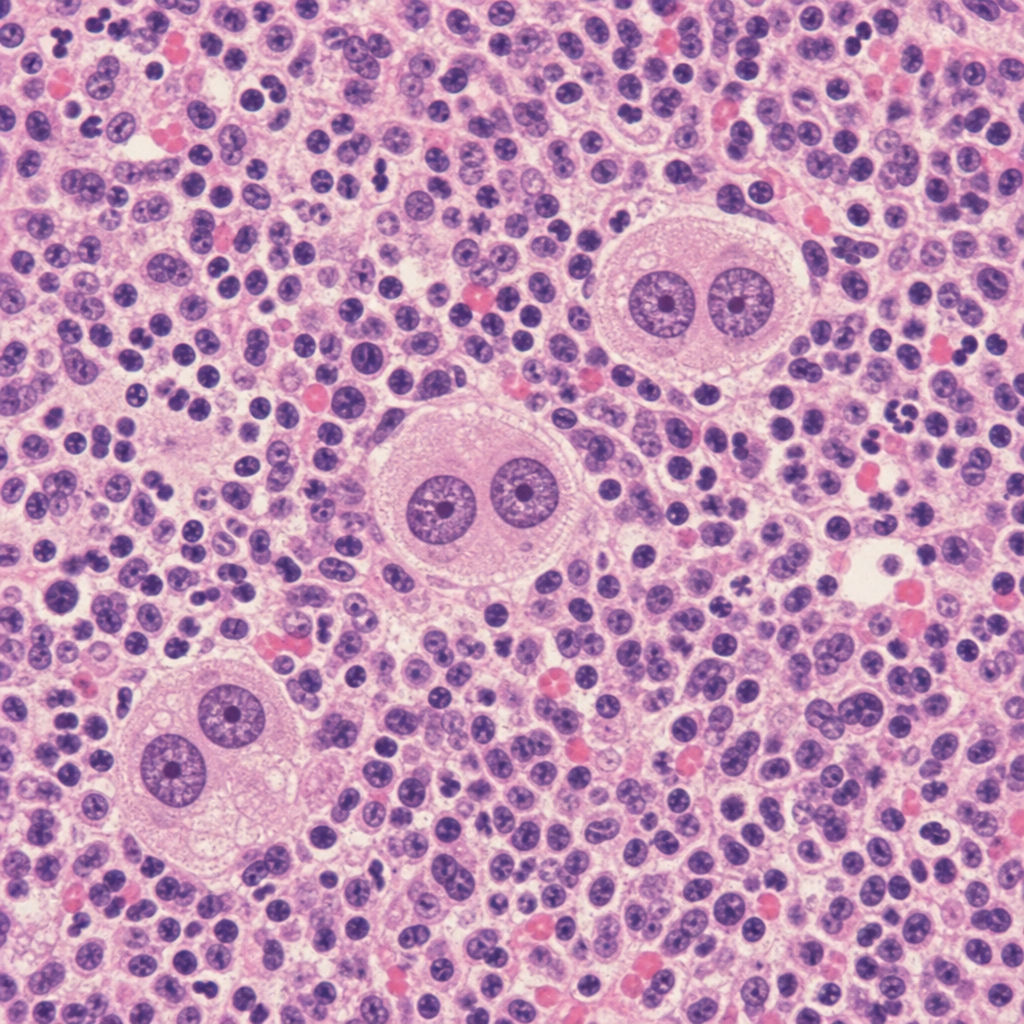
A 34-year-old woman presents with fatigue, night sweats, and a painless 3 cm right cervical lymph node. Excisional biopsy is performed. The photomicrograph demonstrates effacement of normal nodal architecture by a mixed infiltrate of lymphocytes, plasma cells, eosinophils, and neutrophils, within which large binucleated cells with prominent eosinophilic 'owl-eye' nucleoli are identified. Immunohistochemistry on these large cells shows CD15+, CD30+, CD45−, CD20−. A second pathologist reviewing the case suggests an alternative diagnosis based on the presence of numerous eosinophils and a single large mononuclear variant of the same cell type (a classic Reed-Sternberg cell variant within classic Hodgkin lymphoma, not an LP/popcorn cell). Which histological subtype is most consistent with the described immunophenotype and cellular composition?
A 7-year-old girl is brought to her pediatrician by her mother because of puffiness under both eyes in the morning. The mother reports that the child has just recovered from a seasonal influenza infection a few days ago. Vital signs include: temperature 37°C (98.6°F), blood pressure 100/67 mm Hg, and pulse 95/min. On examination, there is facial edema and bilateral 2+ pitting edema over the legs. Laboratory results are shown: Serum albumin 2.1 g/dL Serum triglycerides 200 mg/dL Serum cholesterol 250 mg/dL Urine dipstick 4+ protein Which of the following casts are more likely to be present in this patient’s urine?
A 6-year-old girl comes with her parents to the physician's office to initiate care with a new physician. The patient was recently adopted and her parents do not know her birth history; however, she has had some issues with fatigue. They were told by the adoption agency that the patient has required blood transfusions for "low blood count" in the past but they are not aware of the reason for these transfusions. Her temperature is 37.8°C (99.8°F), blood pressure is 110/84 mmHg, and pulse is 95/min. Physical examination is notable for conjunctival pallor, pale skin, and mild splenomegaly. A complete blood count is taken in the office with the following results: Hemoglobin: 6.8 g/dL Leukocyte count: 5,000/mm^3 Platelet count: 190,000/mm^3 Peripheral smear shows spherocytes and further analysis reveals rigid red blood cells. The most likely cause of this patient's symptoms has which of the following modes of inheritance?
An investigator is studying the immunologic response to a Staphylococcus aureus toxin in a mouse model. Fourteen days after injecting mice with this toxin, he isolates antibodies against neutrophil proteinase 3 in their sera. A patient with high concentrations of these antibodies would most likely present with which of the following clinical features?
An 8-year-old boy has a known genetic condition in which the substitution of thymine for adenine in the 6th codon of the beta globin gene leads to a single-point substitution mutation that results in the production of the amino acid valine in place of glutamic acid. The patient comes to the clinic regularly for blood transfusions. What is the most likely laboratory finding that can be observed in this patient?
A 31-year-old man comes to the emergency department for acute tearing chest pain that radiates to the back. Despite appropriate therapy, the patient dies. Autopsy shows an increase in mucoid extracellular matrix and loss of smooth muscle cell nuclei in the media of large arteries. Which of the following additional findings is most likely in this patient?
Two hours after undergoing allogeneic kidney transplantation for polycystic kidney disease, a 14-year-old girl has lower abdominal pain. Examination shows tenderness to palpation in the area the donor kidney was placed. Ultrasound of the donor kidney shows diffuse tissue edema. Serum creatinine begins to increase and dialysis is initiated. Which of the following is the most likely cause of this patient's symptoms?
A 41-year-old African American woman presents to her primary care physician with a 3-week history of lower extremity edema and shortness of breath. She says that she has also noticed that she gets fatigued more easily and has been gaining weight. Her past medical history is significant for sickle cell disease and HIV infection for which she is currently taking combination therapy. Physical exam is significant for periorbital and lower extremity edema. Laboratory testing is significant for hypoalbuminemia, and urinalysis demonstrates 4+ protein. Which of the following would most likely be seen on kidney biopsy in this patient?
Practice by Chapter
Liver pathology (hepatitis, cirrhosis)
Practice Questions
Gallbladder and biliary tract disorders
Practice Questions
Pancreatic diseases
Practice Questions
Kidney diseases
Practice Questions
Male reproductive pathology
Practice Questions
Female reproductive pathology
Practice Questions
Breast pathology
Practice Questions
Endocrine pathology
Practice Questions
Bone and joint pathology
Practice Questions
Skeletal muscle diseases
Practice Questions
Peripheral nerve disorders
Practice Questions
Soft tissue tumors
Practice Questions
Head and neck pathology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app