
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which of the following statements is false regarding intracellular receptors?
What is the role of oxidation in biotransformation?
Glucuronidation takes place in?
Most variable absorption is seen with which route?
Which of the following drug substrate combinations is incorrectly matched?
Which of the following is NOT an advantage of sustained release preparations over conventional preparations?
Which of the following is the longest acting oral anticoagulant?
A young child weighing 20 kg is administered a drug at a dose of 100 mg/kg body weight. Given that the plasma concentration of the drug is 2 mg/dL and the clearance is 13,860 mL/hr, calculate the time required to reach steady state plasma concentration.
Most common enzyme for drug metabolism and detoxification reactions?
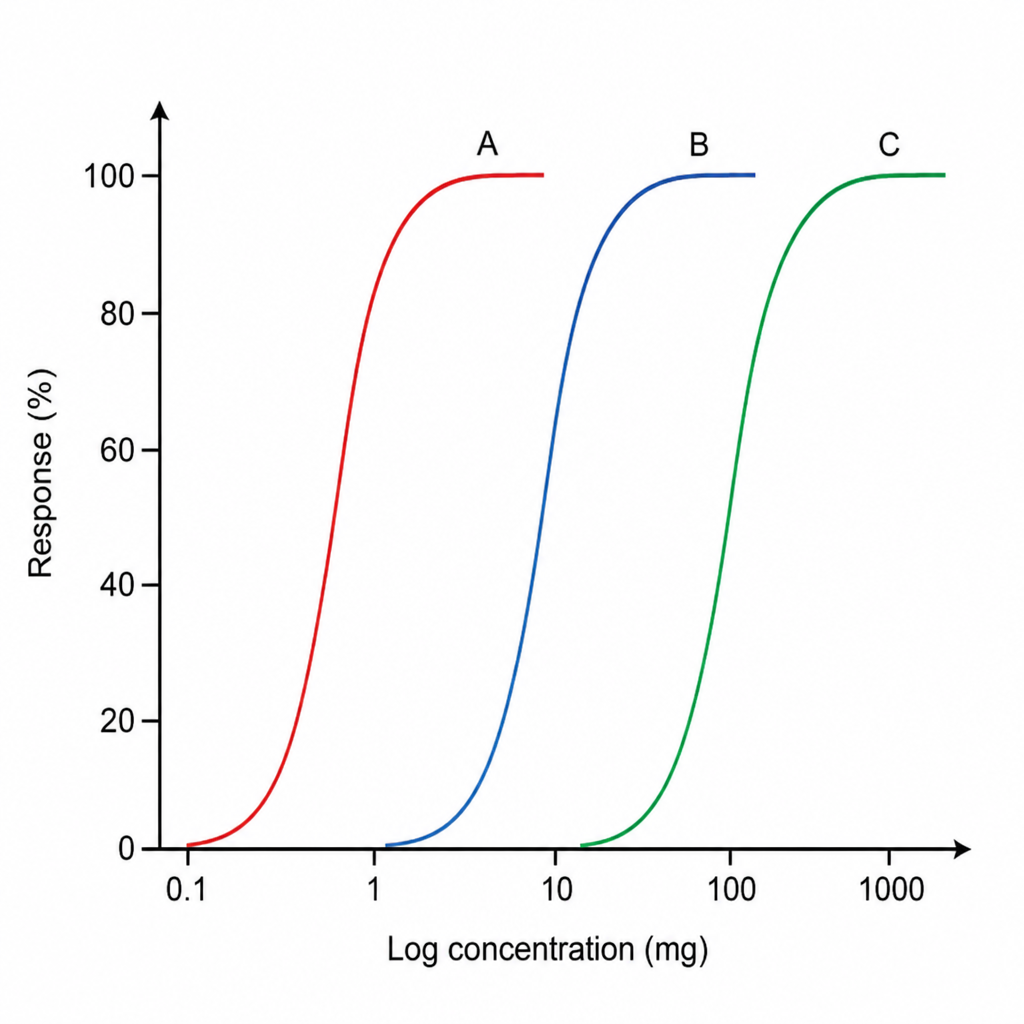
Refer to the graph showing three drugs A, B, and C. Which of the following drugs has the highest potency?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app