
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
What are the purposes of using epinephrine in conjunction with local anesthetics?
A drug has a therapeutic index of 5 and an ED50 of 2 mg. What is the LD50 of the drug?
A new drug shows that its therapeutic dose-response curve and toxic dose-response curve are closely positioned on the dose axis. What can be inferred about its therapeutic index and safety profile?
When comparing oral and intravenous routes of administration, which statement best describes the difference in bioavailability?
A patient requires immediate relief from an acute asthma attack. Which route of administration would provide the fastest effect?
A patient with a severe infection is given a drug with a half-life of 8 hours. How long will it take for the drug's concentration to decrease to 25% of its original value?
Comparing cisplatin and carboplatin, which factor primarily accounts for the difference in nephrotoxicity between these two chemotherapeutic agents?
Which of the following statements is most accurate regarding why fentanyl is preferred over morphine for acute pain management in emergency settings?
In a drug exhibiting first-order kinetics, how would an increase in dose affect the time required to reach the steady-state concentration?
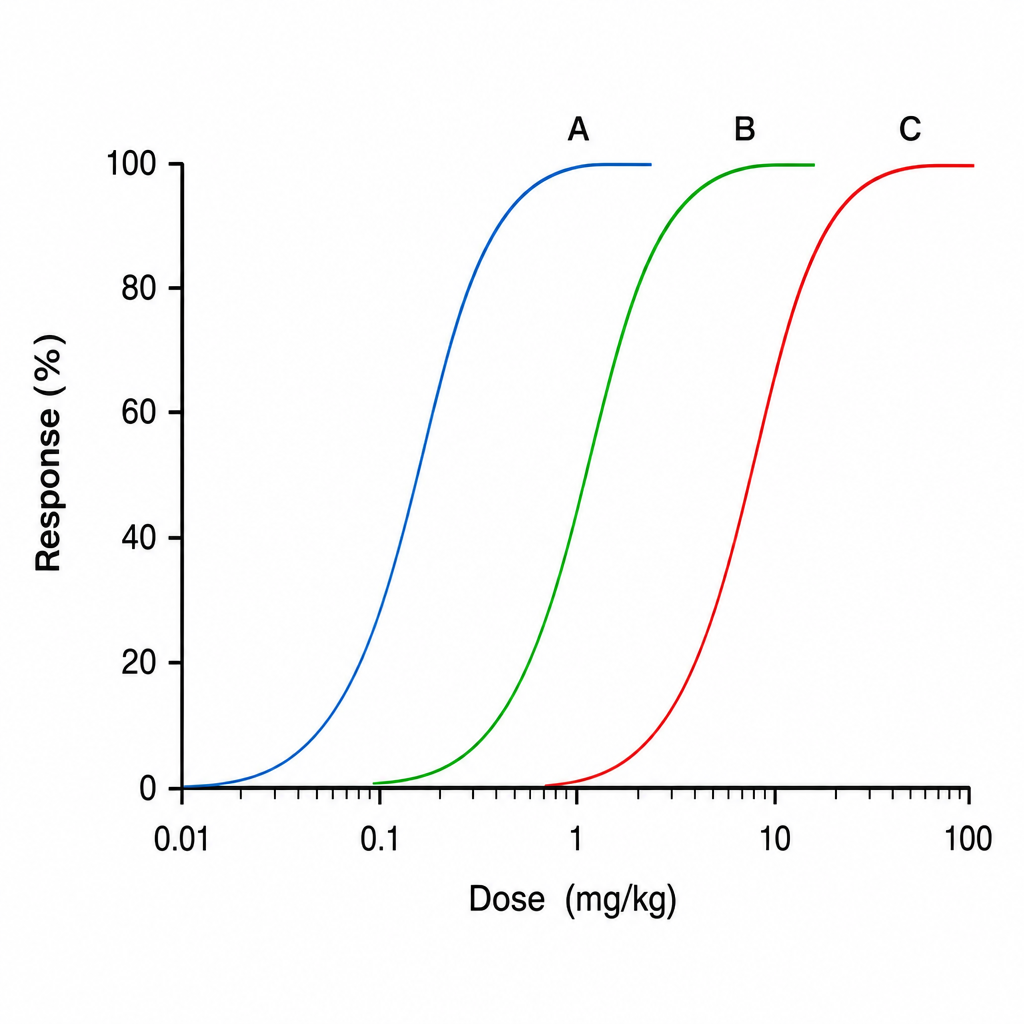
The graph below shows dose-response curves for three drugs A, B, and C. Which of the following drugs has the highest potency?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app