
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which of the following does not penetrate intact skin?
In case of cyanide poisoning, antidote of amyl nitrite is given. This is an example of:
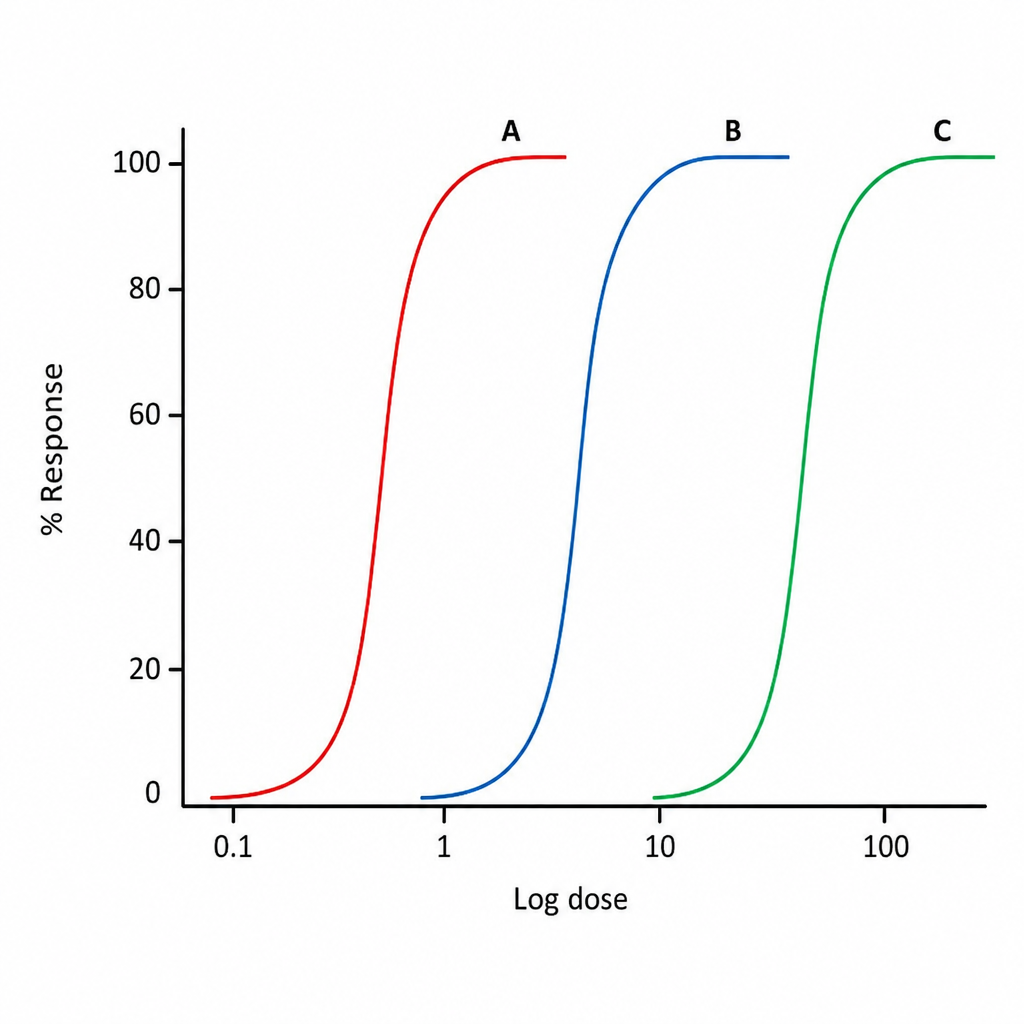
Which of the following drugs shown in the graph below has highest potency?
Longest acting local anesthetic agent is -
Low apparent volume of distribution of drug indicates that:
Duration of action of proparacaine:
G-protein coupled receptor that does not act through opening of potassium channels is:
All are true for cytochrome P450 enzymes EXCEPT:
What is the primary role of Cytochrome P450 enzymes in the liver?
Which of the following statements is true regarding zero-order kinetics?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app