
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
A U-shaped dose-response curve is typically observed with which of the following?
When a drug binds to a receptor and causes an action opposite to that of an agonist, what is this called?
When a drug binds to a receptor and causes an action opposite to that of an agonist, it is called as:
The pharmacokinetic properties of a new drug are being studied in normal volunteers during phase I clinical trials. The volume of distribution and clearance determined in the first subject are 40 L and 2.0 L/hour, respectively. What is the approximate half-life of the drug in this subject?
Which of the following statements regarding Cytochrome P450 is FALSE?
Which of the following is an ionotropic receptor?
A patient with normal renal function has received a daily maintenance dose of digoxin for 2 weeks. If the dosage is changed, in approximately how long would the new steady-state plasma digoxin concentration be achieved?
Exogenous adrenaline is metabolized by:
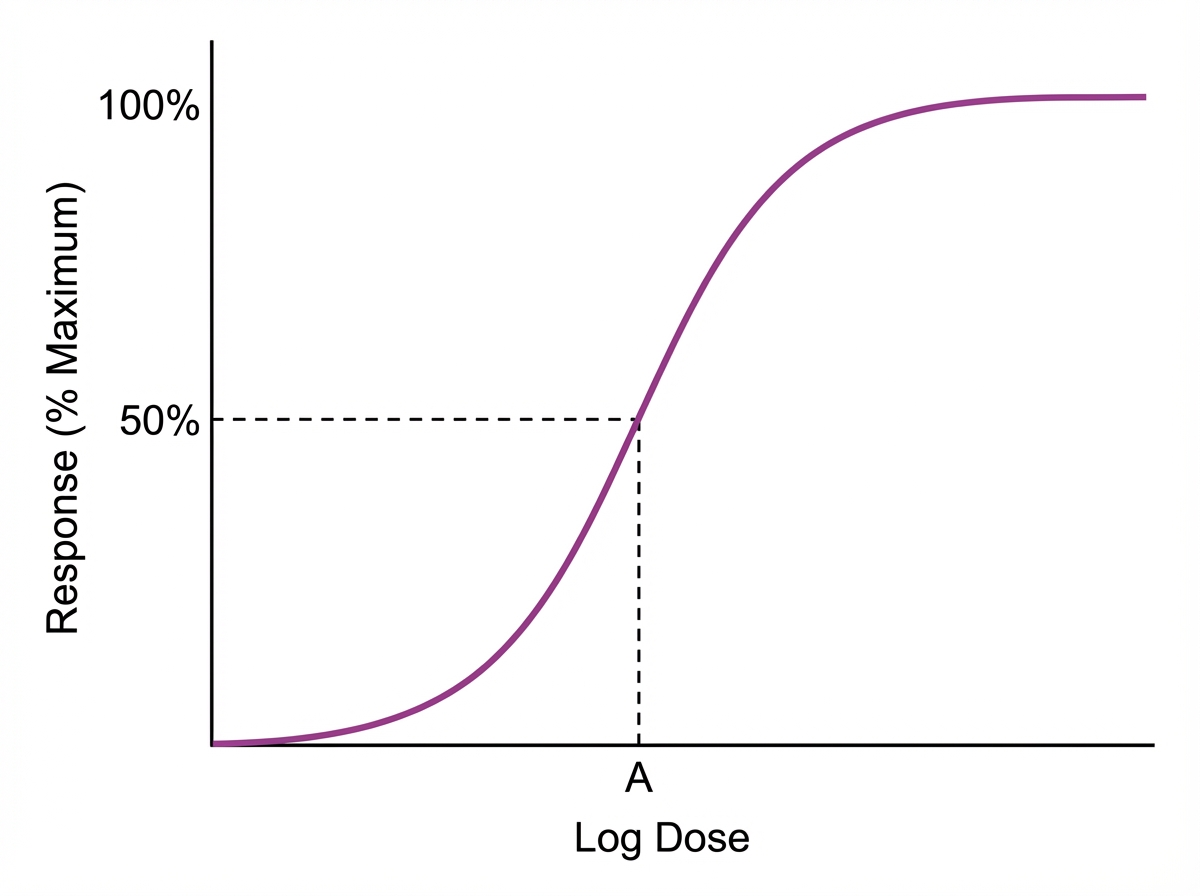
In the given pharmacodynamic curve, point A corresponds to:
Receptor-mediated action is not seen in which of the following?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app