
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
The following plot of log dose of norepinephrine on X-axis and response in the form of increase in cardiac contractility on Y-axis represents which type of dose-response relationship based on the nature of the response measured?
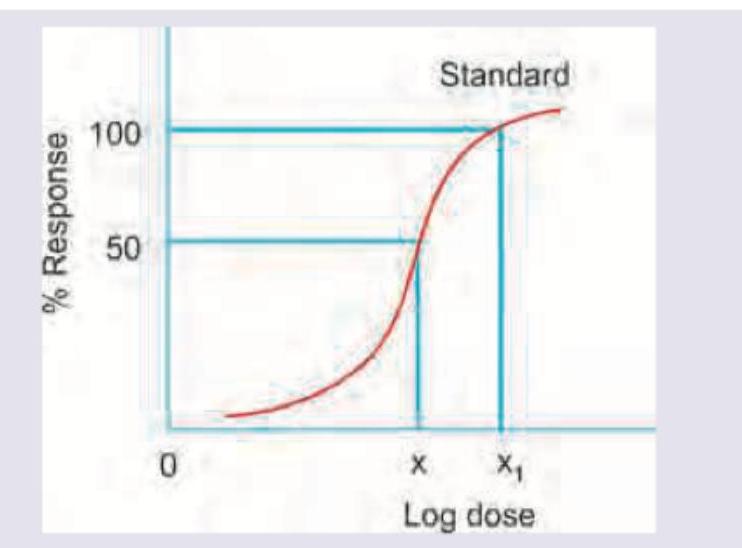
Which of the following statement is correct regarding the given DRC? (AllMS Nov 2016)
The maximum safe dose for Lignocaine (without adrenaline) as a local anaesthetic drug is :
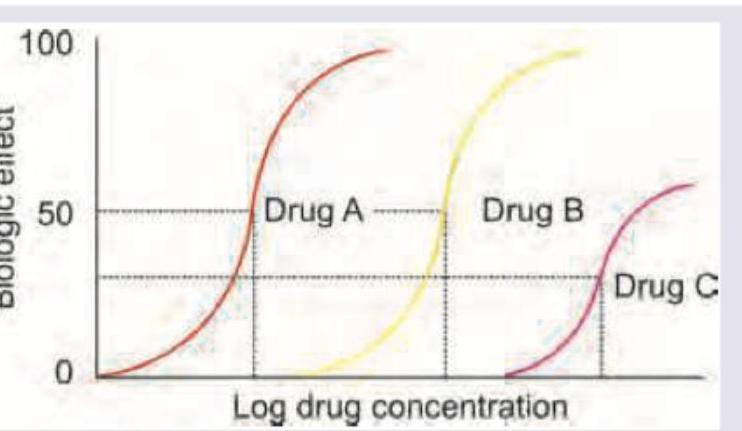
Which of the following statements is correct regarding the given graph?
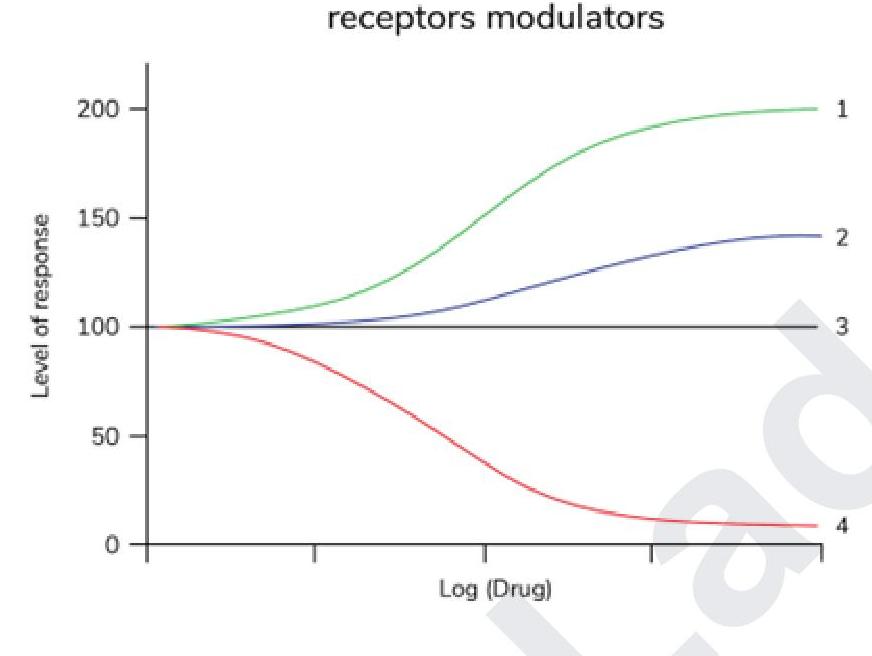
Which of the following is true about clinical therapeutic index?
Choose the correct options regarding the route of administration and bioavailability. A- Intravenous =1 B- 0.75< Oral <1 C-0.75 <IM ≤ 1 D- 0.75<SC ≤ 1 IM - Intramuscular SC- Subcutaneous
What would happen to the half-life and plasma concentration of a drug which follows first-order kinetics, if the dose is doubled?
Find therapeutic index of drug from the information given below in the graph
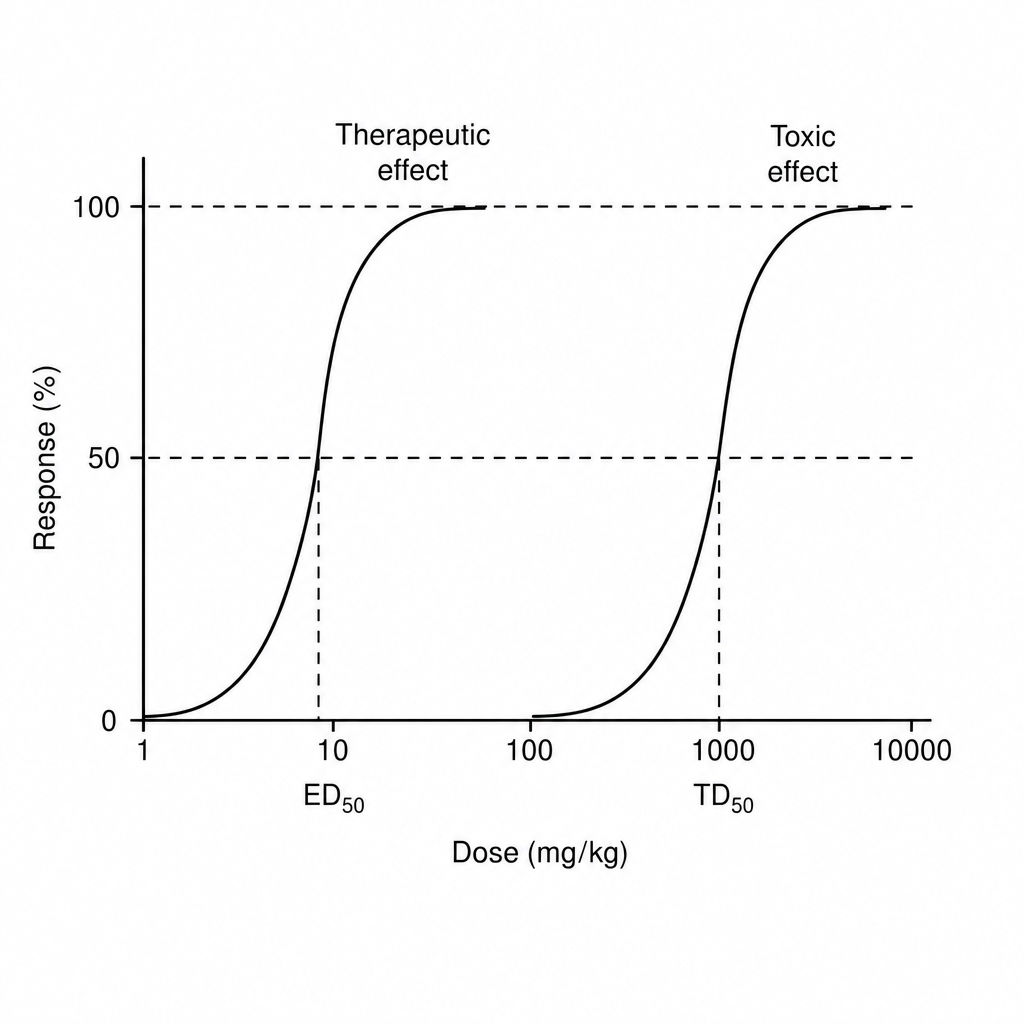
Therapeutic index of a drug is an indicator of:-
Pharmacodynamics deals with:-
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app