
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which of the following statements about biotransformation is NOT true?
A 75 kg person is administered a drug with a half-life of 3 hours. What is the approximate clearance of the drug?
Which statement best describes the relationship between drug potency and efficacy in dose-response curves?
Loading dose of an oral drug depends on all of the following except?
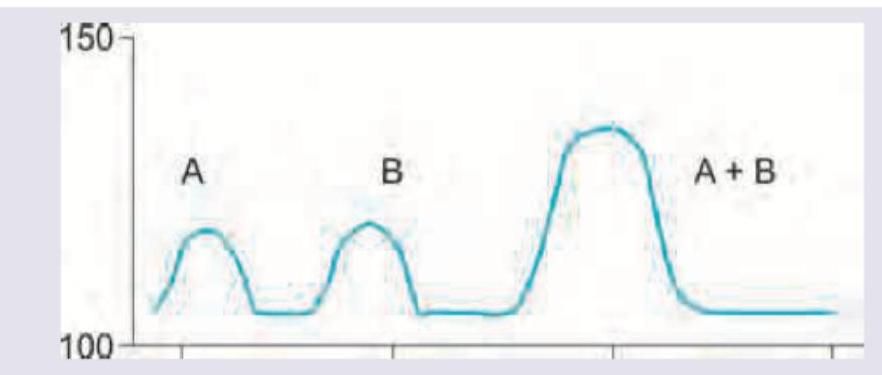
The effect of both drugs given together on the blood pressure of a patient was evaluated in comparison with the effect of individual drugs on BP. The following curve represents: (Recent NEET Pattern 2016-17)
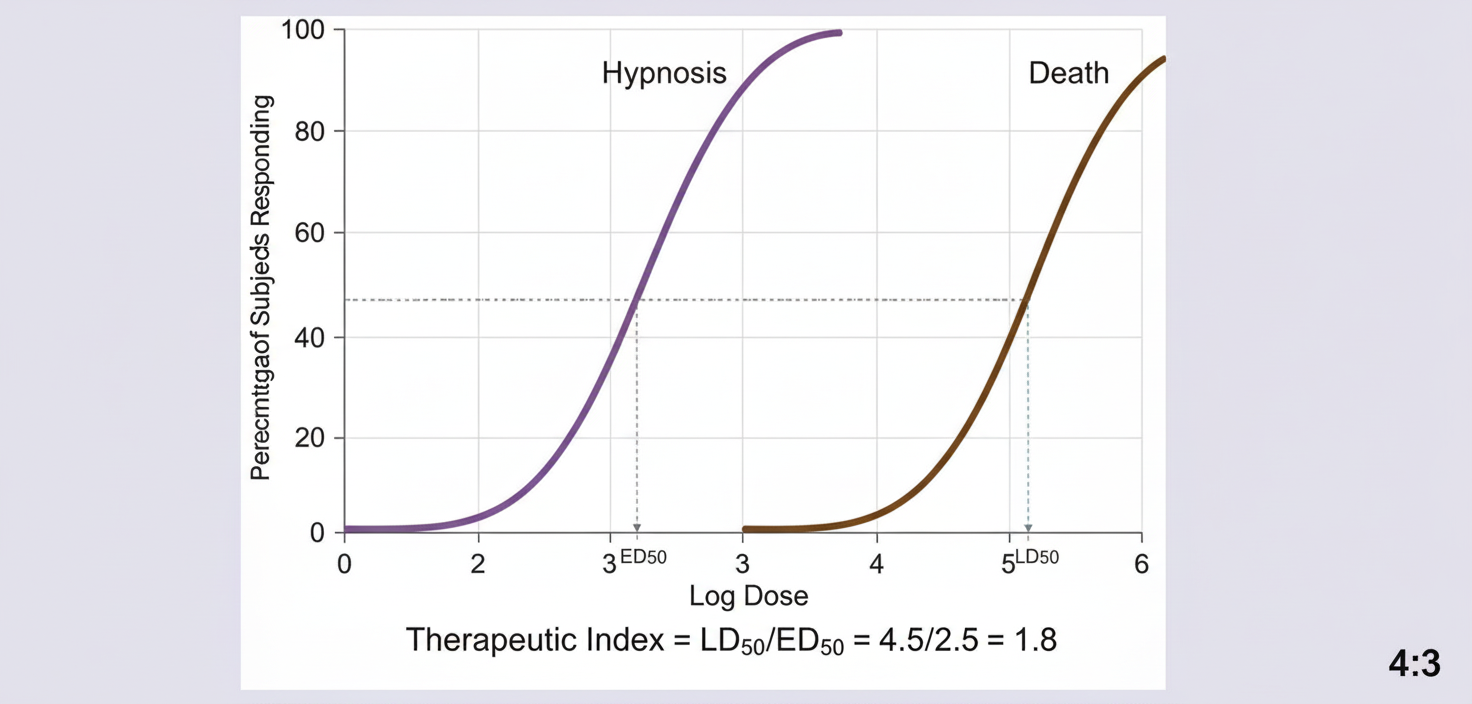
A new sedative-hypnotic drug is being evaluated in preclinical toxicity studies. The following quantal dose-response curve is obtained showing the hypnotic effect and lethal effect. Calculate the therapeutic index of the drug being tested.
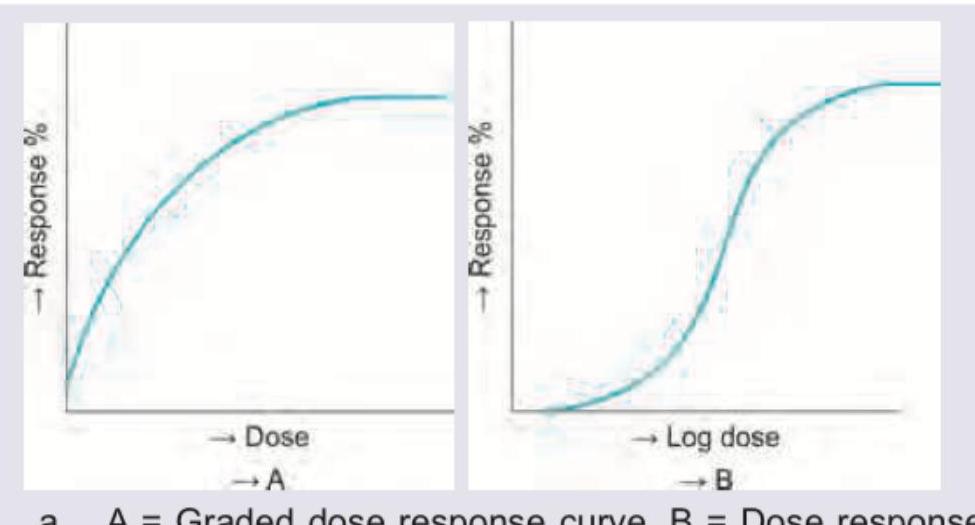
Which is correct about the curves shown?
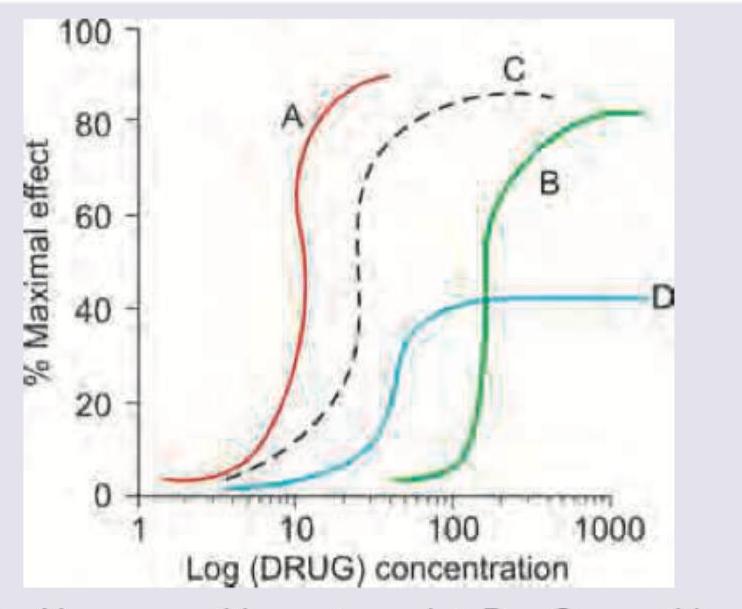
The following curve shows a graded dose-response curve. Drug A and drug B are bronchodilators. What do drug C and D represent?
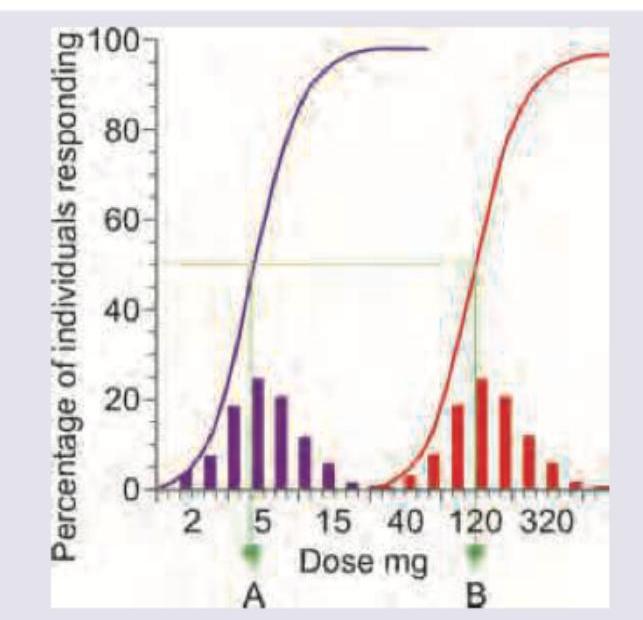
What do A and B represent in the curve shown below?
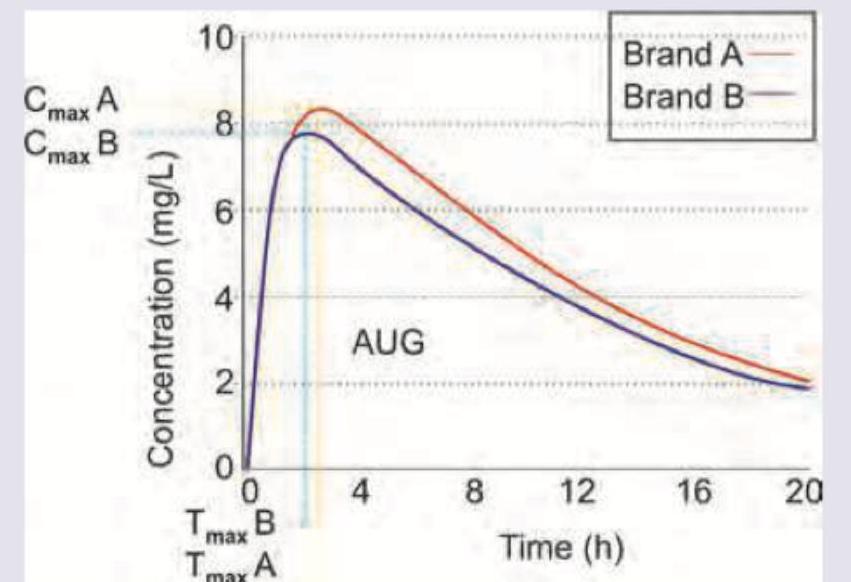
Brand A of liposomal amphotericin B is of innovator company and brand B is of a generic company. AUC of Brand A is 124 mg.h/L and AUC of brand B is 115 mg.h/L. Which of the following statements is correct?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app