
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Urinary alkalinizing agents are administered in case of poisoning due to which type of drugs?
Which of the following drug combinations represents psychological antagonism?
Which of the following characteristics is associated with a high volume of distribution for a drug?
An elderly patient presents with myocardial infarction and a severe ventricular arrhythmia. The chosen antiarrhythmic drug has a narrow therapeutic window, with a minimum toxic plasma concentration 1.5 times the minimum therapeutic plasma concentration. The drug's half-life is 6 hours. It is crucial to maintain the plasma concentration above the minimum therapeutic level to prevent lethal arrhythmia. Which of the following dosing regimens is most appropriate?
Which of the following is an ionic channel?
Which is the most common cytochrome P450 enzyme associated with drug metabolism?
The ratio of the median lethal dose to the median effective dose is the:
True regarding the following is?
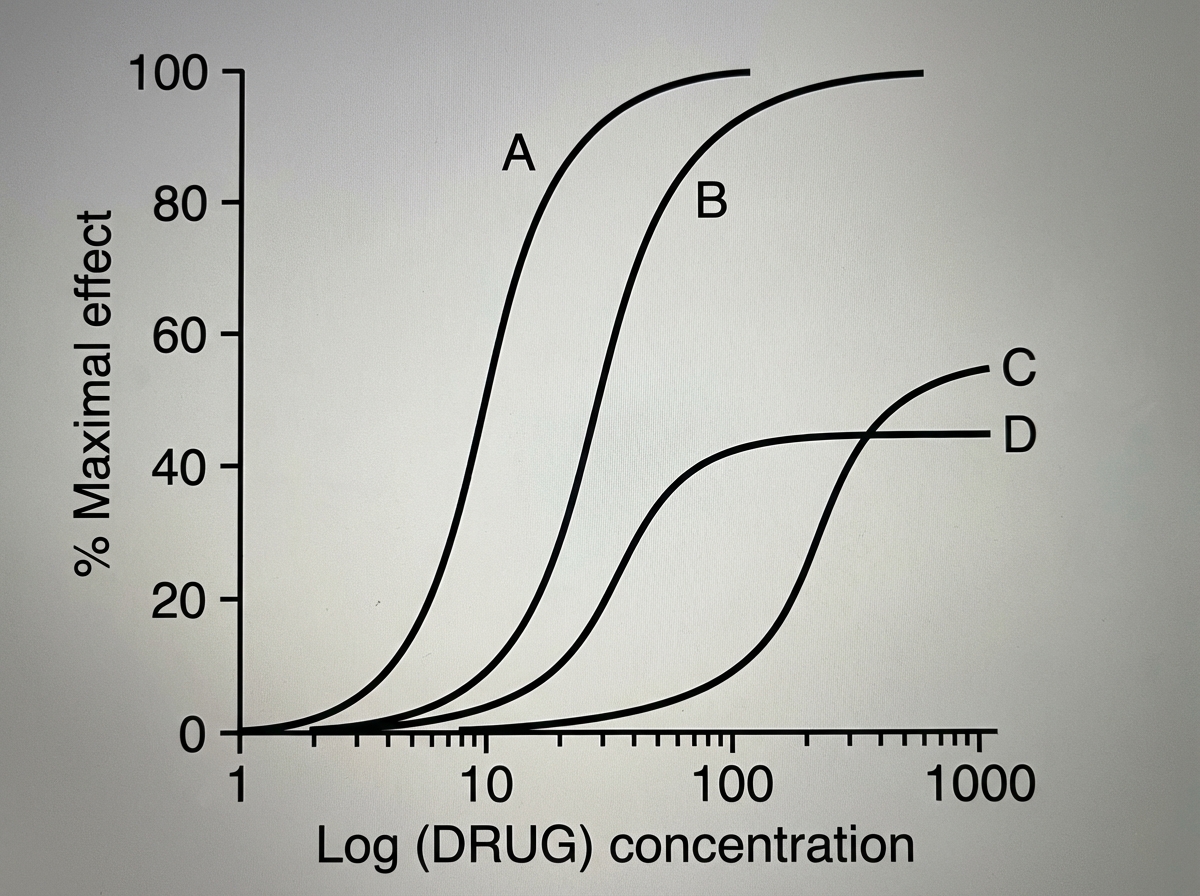
Regarding phenytoin, all are true except?
All of the following drugs act on ionic channels except?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app