
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which of the following is characteristic of drugs exhibiting zero-order kinetics of elimination?
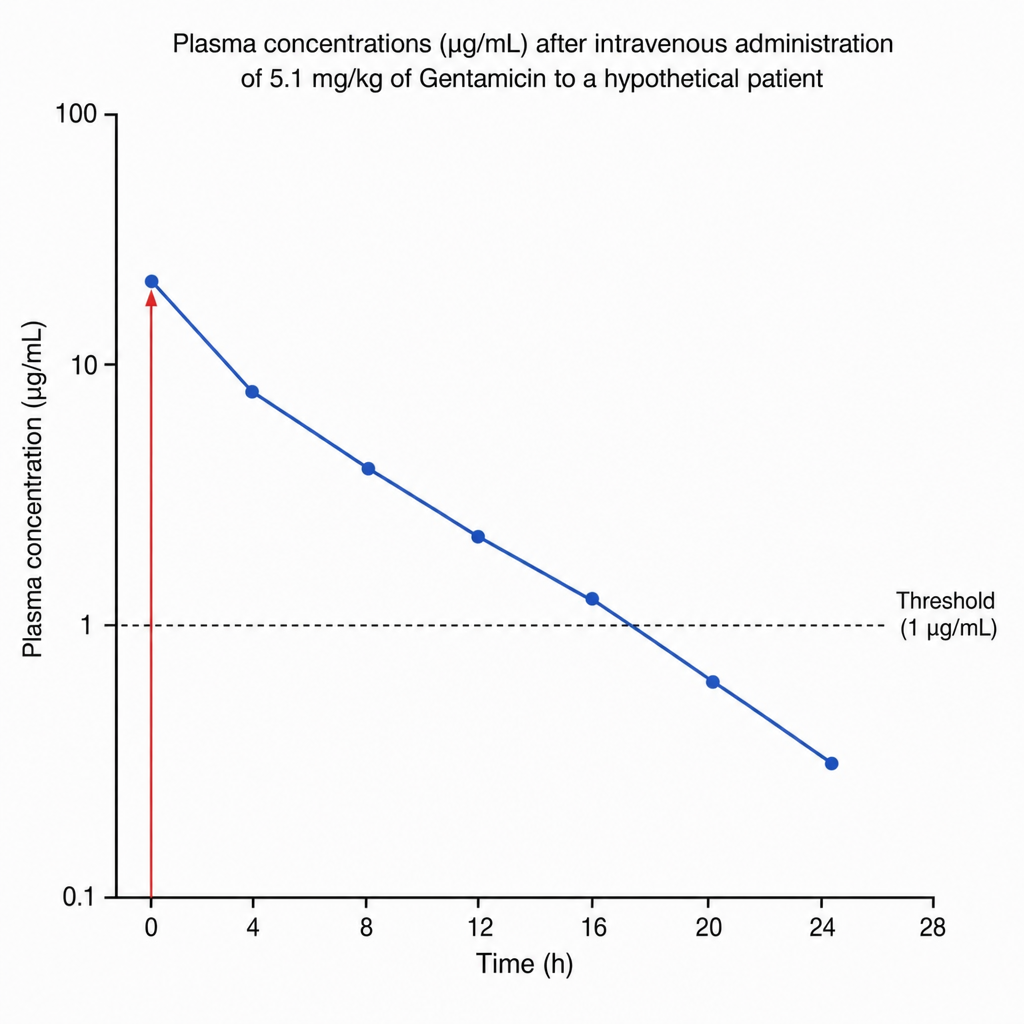
Plasma concentrations (µg/mL) after intravenous administration of 5.1 mg/kg of Gentamicin to a hypothetical patient are depicted in the graph. Based on the concentration-time curve, what is the approximate elimination half-life of Gentamicin in this patient?
Which drug is highly distributed to body fat?
Which of the following properties of a drug will enable it to be used in low concentration?
Procainamide infusion is initiated. Its half-life is 2 hours. The infusion begins at 9:00 a.m. and ends at 1:00 p.m. on the same day. At 1:00 p.m., the blood concentration is found to be 3 mg/L. What is the probable steady-state concentration after 2 days of infusion?
Loading dose primarily depends on which pharmacokinetic parameter?
Arrange the following drugs according to their half-life in increasing order: 1. Amiodarone 2. Adenosine 3. Esmolol 4. Omeprazole
Which of the following medications inhibits the effect of oral contraceptive pills?
A 500-mg dose of a drug has therapeutic efficacy for 6 hours. If the half-life of the drug is 8 hours, for how long would a 1-gm dose be effective?
All of the following are examples of mechanism-based inhibition, EXCEPT?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app