
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which pathological state alters the volume of distribution of many drugs?
Which of the following terms best describes a drug that blocks the action of adrenaline at its receptors by occupying those receptors without activating them?
Which of the following statements about biotransformation is untrue?
Which of the following drugs has the least oral bioavailability?
The time required to reach the steady state after a dosage regimen depends on which of the following?
In drug metabolism, what is the primary role of the hepatic cytochrome P-450 system?
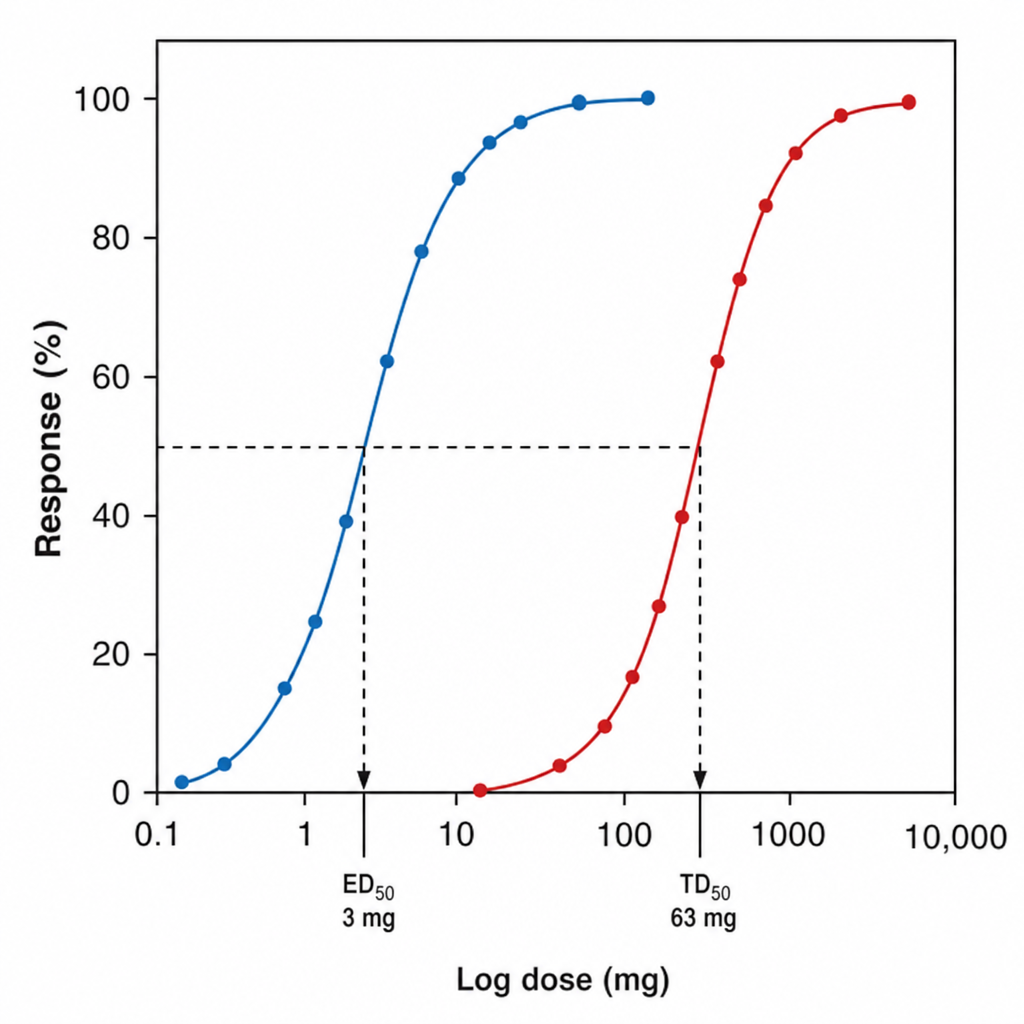
What is the therapeutic index of the drug based on the provided dose-response curve (DRC)?
The pharmacokinetics of Paroxetine include its ability to be easily absorbed after oral administration in healthy volunteers even after food is ingested. Which of the following is TRUE regarding Paroxetine's pharmacokinetics?
Which of the following most accurately describes the transmembrane signaling process involved in the steroid hormone action?
All of the following statements regarding volume of distribution are true except?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app