
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which of the following does NOT induce microsomal enzymes?
When atrial fibrillation persists despite digoxin therapy, an inadequate digoxin dose is suspected. Plasma levels of digoxin for confirmation are typically drawn after a certain interval following the last dose. Considering pharmacokinetic principles, which factor is most crucial for determining this appropriate sampling time?
At 12 hours after intravenous administration of a bolus dose, the plasma level of a drug is 3 mg/L. If the volume of distribution (Vd) is 10 L and the elimination half-life is 6 hours, what was the dose administered?
Which of the following is NOT a type of oxidative drug metabolism?
A drug has 40% absorption and a hepatic extraction ratio of 0.6. What is the bioavailability of the drug?
Which of the following is false regarding spare receptors?
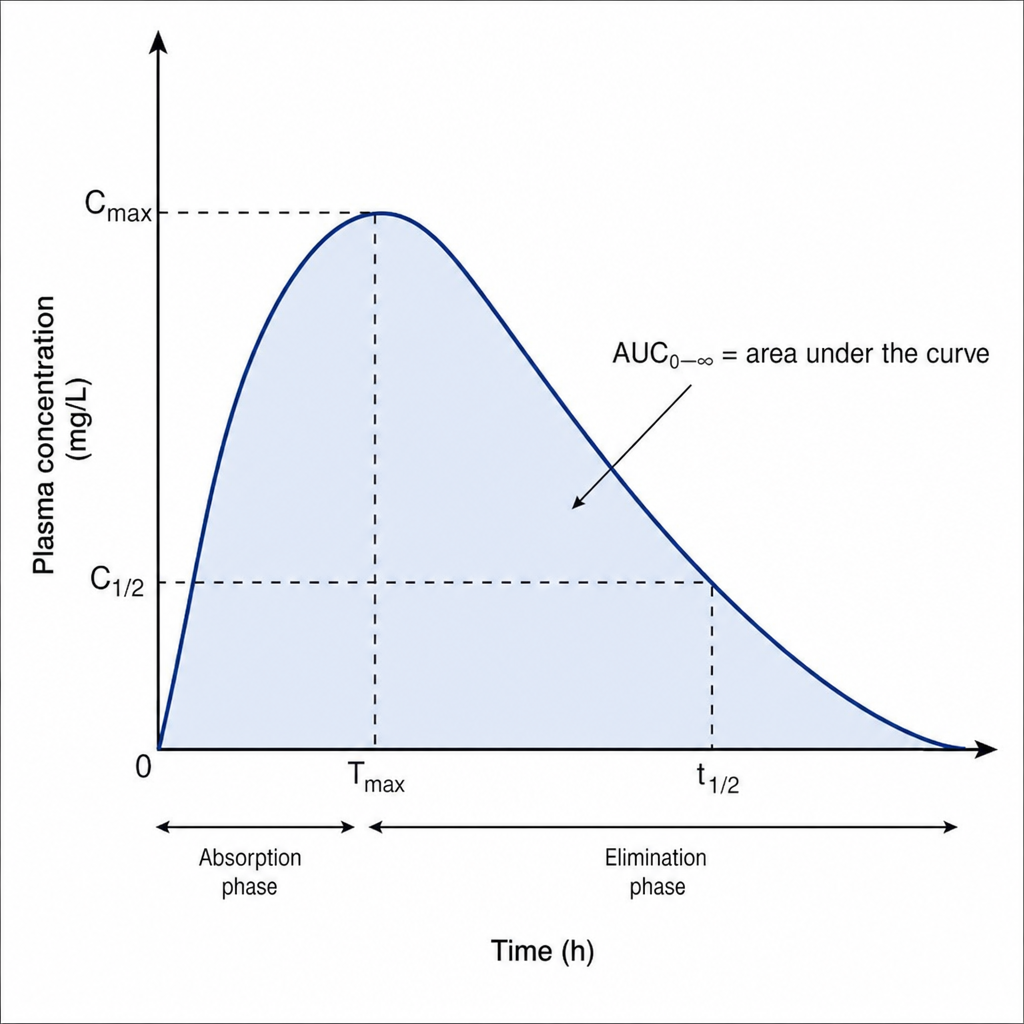
What does the plasma concentration versus time graph indicate?
Phase II drug metabolism reactions include all of the following EXCEPT:
A new antifungal medication has a half-life of 6 hours. If a continuous intravenous infusion of this drug were started, how long would it take to reach 75% of steady state?
Loading dose is given for which of the following drugs?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app