
Pharmacokinetics and Pharmacodynamics — MCQs

On this page
Which immunosuppressive medication blocks the effects of interleukin -2 (IL-2) without inhibiting calcineurin activity?
A novel anti-aging compound induces autophagy by inhibiting EP300 histone acetyltransferase, leading to deacetylation of autophagy-related proteins. Which known anti-aging agent works by the same mechanism?
Which of the following is correctly matched (Anti-aging compound - Molecular pathway)?
A 45-year-old chronic smoker is being counselled for smoking cessation therapy. Which of the following statements regarding nicotine replacement and pharmacotherapy is correct?
Identify the type of elimination kinetics represented in the given graph:
Calculate the Therapeutic Index of the given drug from the dose-response curve below:
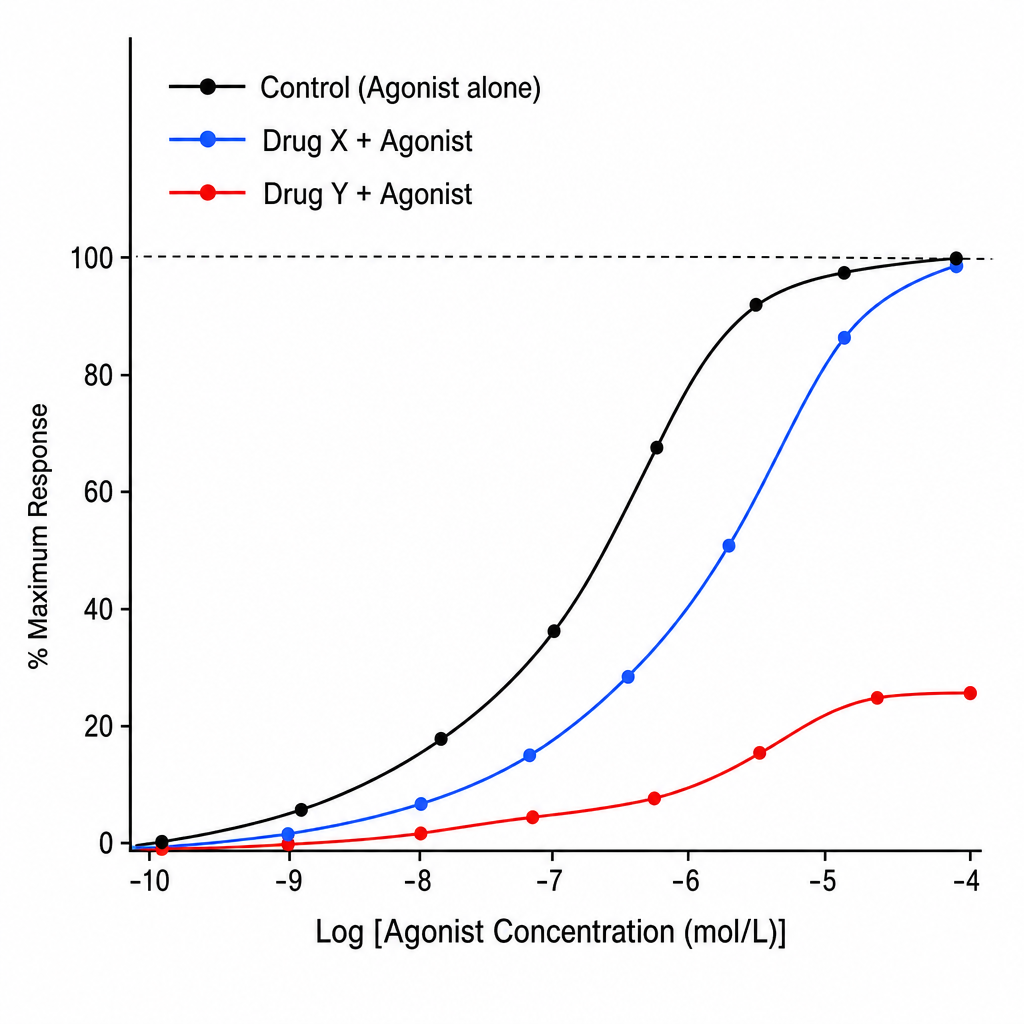
A pharmacology researcher is studying the interaction of a new drug with a receptor system. Two candidate antagonist compounds (Drug X and Drug Y) are tested against a standard agonist at increasing concentrations. The resulting dose-response curves are plotted (Image 1). In the graph, the X-axis represents Log [Agonist Concentration (mol/L)] and the Y-axis represents % Maximum Response (0–100%). Three labeled sigmoid curves are shown: (1) 'Control' — the agonist alone, reaching 100% Emax; (2) 'Drug X + Agonist' — a parallel rightward shift of the control curve with unchanged Emax (~100%); (3) 'Drug Y + Agonist' — a rightward shift with a clearly depressed/lowered Emax (~50–60%). Based on the curves, which of the following statements best describes the mechanism of Drug Y?
Zero order kinetics is followed by all of the following drugs EXCEPT?
If a drug is excreted in urine at the rate of 10 mg/hr at a steady-state plasma concentration of 5 mg/L, then its renal clearance is:
A drug with a plasma half-life of 12 hours is administered twice a day. Steady-state plasma concentration reached is 300 mg/dl. Which of the following statements about this drug is not true?
Practice by Chapter
Absorption and Bioavailability
Practice Questions
Drug Distribution and Protein Binding
Practice Questions
Biotransformation and Metabolism Pathways
Practice Questions
Renal and Non-renal Excretion
Practice Questions
Compartment Models
Practice Questions
Dose-Response Relationships
Practice Questions
Drug Efficacy and Potency
Practice Questions
Drug Tolerance and Tachyphylaxis
Practice Questions
Population Pharmacokinetics
Practice Questions
Pharmacokinetic Variability
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app