
Neurology — MCQs

On this page
A child presented with clinical features of demyelination. The chances of progression to Multiple Sclerosis is least with which of the following?
The clinical manifestation of Sydenham chorea includes all except?
Which of the following is true regarding Gower sign?
MacEwen sign is seen in which condition?
All of the following are true about meningocele except?
A 3-year-old boy presents with a several-month history of increasing difficulty walking. His parents note an increased inward curvature of the lower spine during ambulation and a more waddling gait. Physical examination reveals these findings, along with enlarged calves. What is the most likely diagnosis?
Which of the following conditions is associated with early complete fusion of all of the cranial sutures?
In which of the following conditions is the given appearance of calves typically observed?
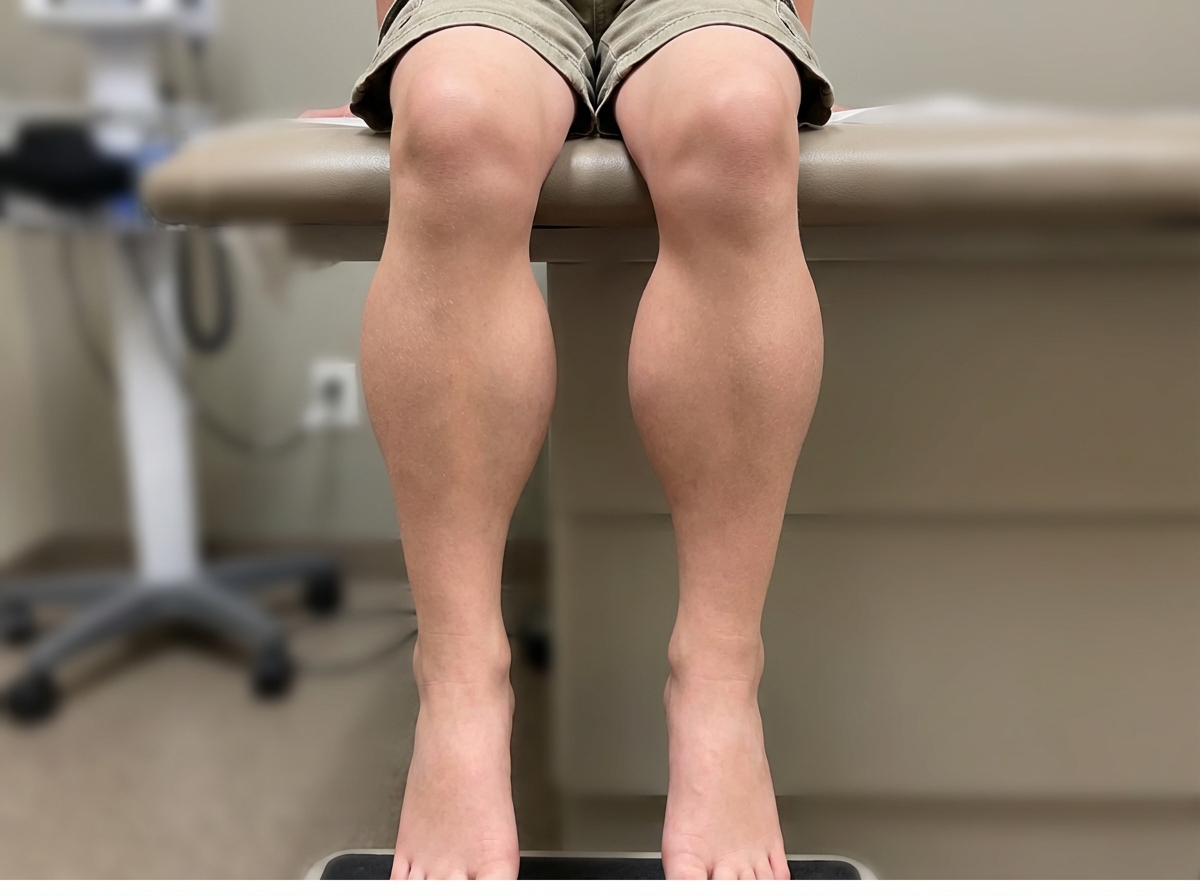
A 10-year-old boy, who was apparently normal at birth, had delayed walking and difficulty in walking. As years passed, weakness increased, and he is now wheelchair-bound. What is your diagnosis?
Neurofibromatosis type 1 is most commonly associated with which of the following tumors?
Practice by Chapter
Seizure Disorders and Epilepsy
Practice Questions
Febrile Seizures
Practice Questions
Headache Disorders
Practice Questions
Cerebral Palsy
Practice Questions
Neural Tube Defects
Practice Questions
Neuromuscular Disorders
Practice Questions
Neurodegenerative Disorders
Practice Questions
CNS Infections
Practice Questions
Hydrocephalus
Practice Questions
Movement Disorders
Practice Questions
Traumatic Brain Injury
Practice Questions
Neuroimaging in Pediatrics
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app