
Neurology — MCQs

On this page
Agenesis of corpus callosum with retinal colobomas and intellectual disability is seen in which condition?
A boy presents with weakness in the lower limbs, calf hypertrophy, a positive Gower's sign, and an elevated CPK value of 10,000. What is the most likely diagnosis?
What is the preferred treatment for Rolandic epilepsy?
A CT scan of a child presenting with CNS pathology reveals a subdural effusion. Which of the following conditions commonly leads to subdural effusion in children?
Comment on the diagnosis of a 1-year-old child?
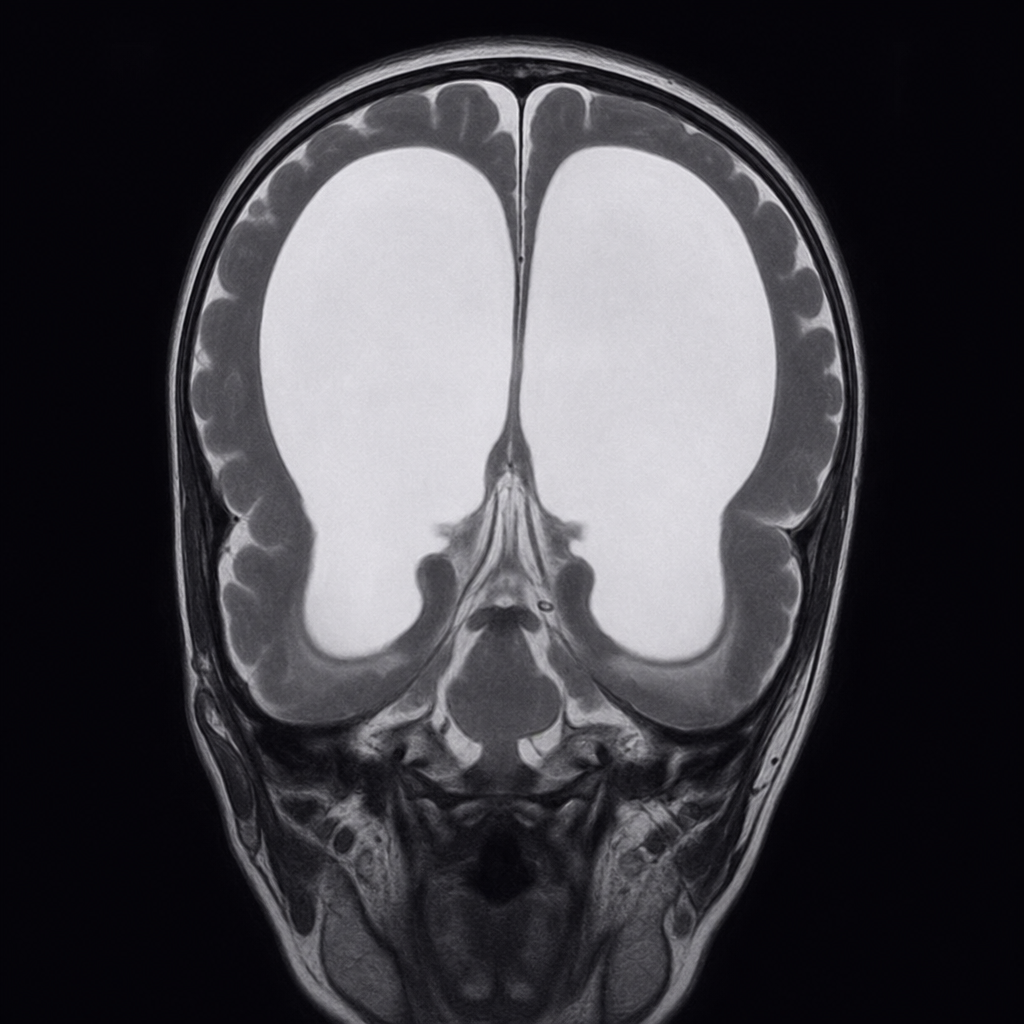
Regarding Dandy-Walker syndrome, which of the following statements are true and which are false? 1. It is the most common posterior fossa malformation. 2. It consists of a cystic expansion of the 4th ventricle in the posterior fossa and midline cerebellar hypoplasia. 3. The classic triad includes hypoplasia of the vermis, cephalad rotation of the vermian remnant, and cystic dilatation of the 4th ventricle extending posteriorly with a small posterior fossa. 4. The most common manifestation is macrocephaly. 5. Management involves shunting the cystic cavity.
Which of the following is NOT typically found in cerebral palsy?
What is the drug of choice for infantile spasms in a child with Tuberous sclerosis?
An infant of 5 months presents with sudden dropping of head and flexion of arms. On examination, the infant has a hypopigmented macule on the trunk. What is the drug of choice for this condition?
All of the following are complications of Duchenne Muscular dystrophy except?
Practice by Chapter
Seizure Disorders and Epilepsy
Practice Questions
Febrile Seizures
Practice Questions
Headache Disorders
Practice Questions
Cerebral Palsy
Practice Questions
Neural Tube Defects
Practice Questions
Neuromuscular Disorders
Practice Questions
Neurodegenerative Disorders
Practice Questions
CNS Infections
Practice Questions
Hydrocephalus
Practice Questions
Movement Disorders
Practice Questions
Traumatic Brain Injury
Practice Questions
Neuroimaging in Pediatrics
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app