
Neurology — MCQs

On this page
A patient presents with intractable convulsions, mental defect, and a facial nevus. What is the most likely diagnosis?
A 4-month-old child presents with excessive irritability and crying, unexplained hyperpyrexia, vomiting, and difficulty feeding for the last 15 days. On admission, the child has rigidity and visual inattentiveness. A CT scan of the brain shows specific findings. What is the probable diagnosis?
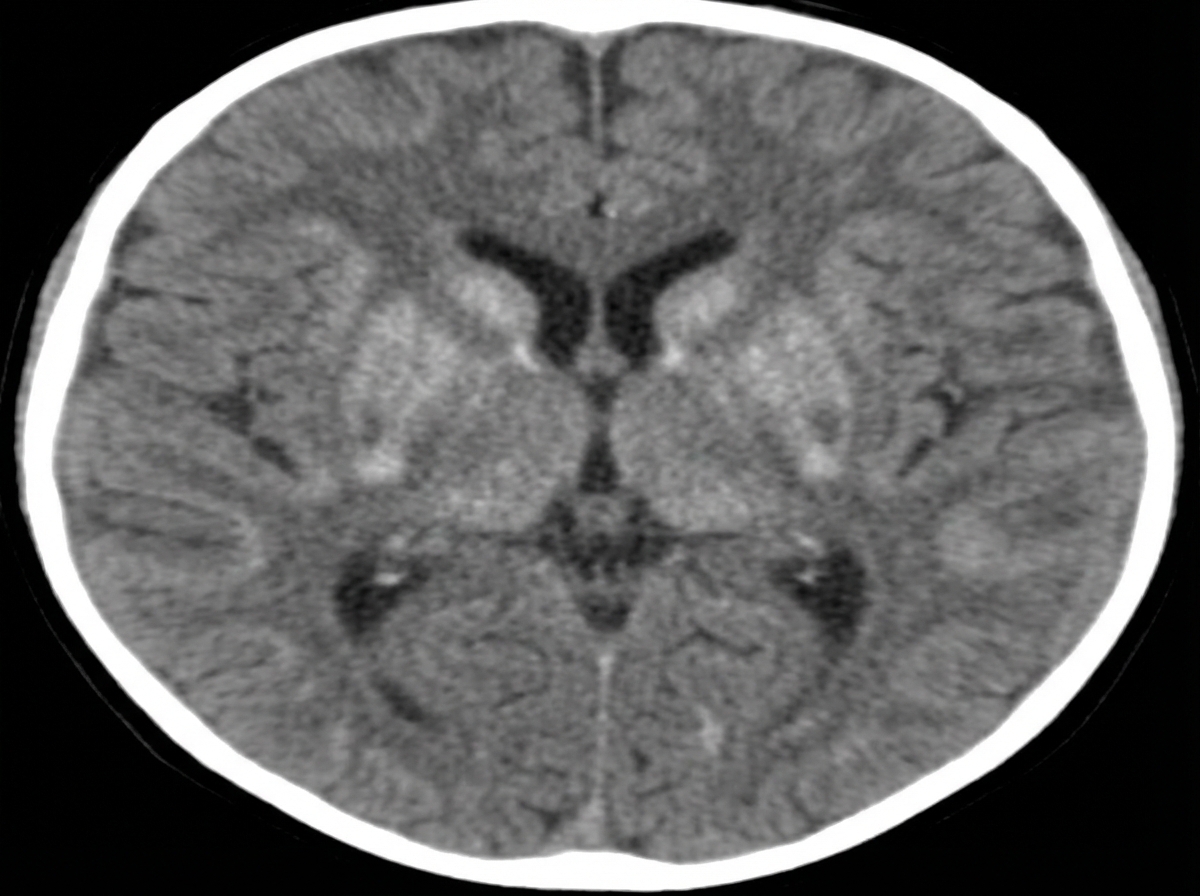
What is the drug of choice for treating myoclonus in children?
Hypsarrhythmia in a child is typically associated with which type of epilepsy?
What is the most probable cause of a large head in this child?
A child presents with unilateral facial pigmentation and a history of multiple seizures. Radiography of the skull revealed a specific finding. What is the most likely underlying diagnosis?
All of the following predominantly involve the white matter EXCEPT?
An 8-year-old child with a history of GTCS came with an episode of convulsions for more than 45 minutes. What will be the appropriate management for this patient?
The infant is being evaluated for recurrent episodes of seizures. EEG shows Hypsarrhythmia. Which drug is preferred for management?
A neonate is found to have multiple hypopigmented macules and ash-leaf spots on Wood's lamp examination. Which of the following is the most likely diagnosis?
Practice by Chapter
Seizure Disorders and Epilepsy
Practice Questions
Febrile Seizures
Practice Questions
Headache Disorders
Practice Questions
Cerebral Palsy
Practice Questions
Neural Tube Defects
Practice Questions
Neuromuscular Disorders
Practice Questions
Neurodegenerative Disorders
Practice Questions
CNS Infections
Practice Questions
Hydrocephalus
Practice Questions
Movement Disorders
Practice Questions
Traumatic Brain Injury
Practice Questions
Neuroimaging in Pediatrics
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app