
Rheumatology and Immunology — MCQs

On this page
Most strongly associated with rheumatoid arthritis among the following is?
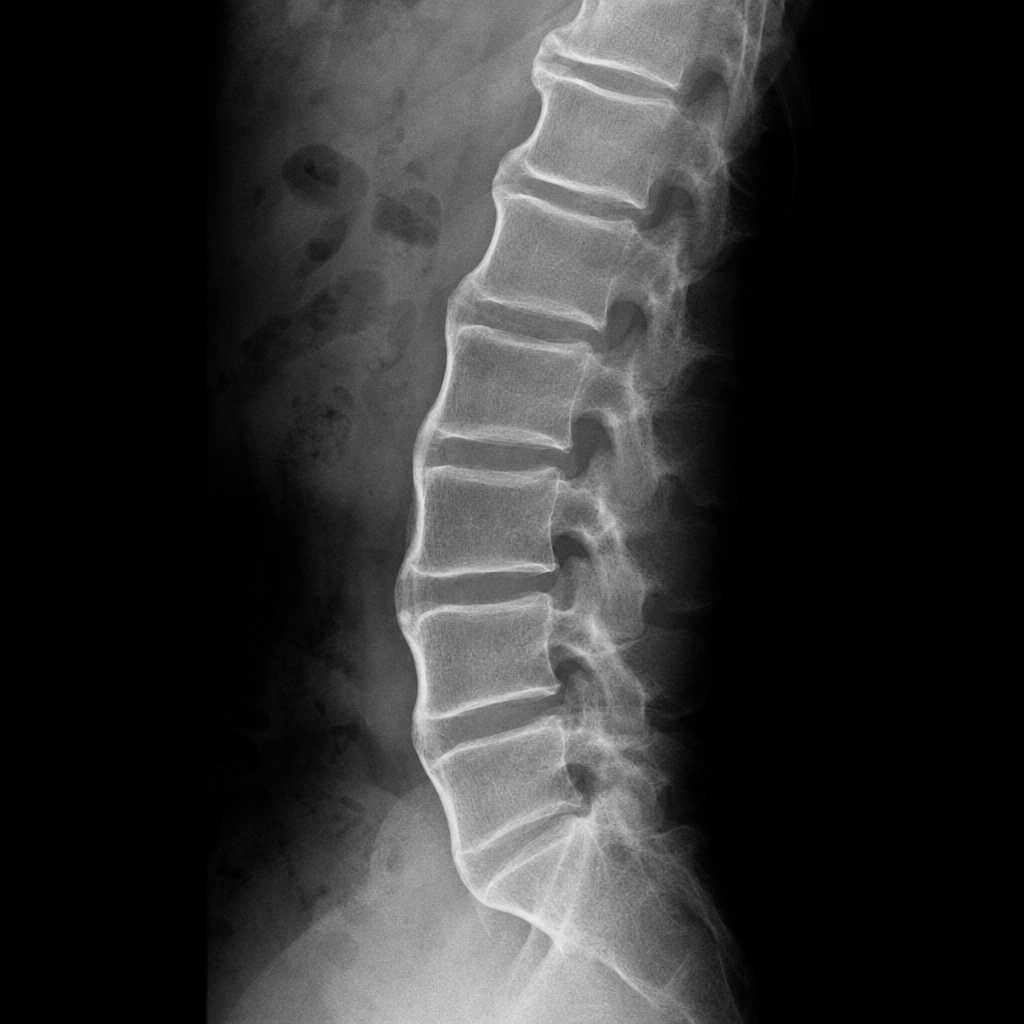
In a middle aged male having back pain, flowing anterolateral ossification bridging at least four contiguous vertebral bodies with preserved disc spaces is seen on Xray. The patient has -
In gout tophi are not seen in?
Patient following peanut consumption presented with laryngeal edema, stridor, hoarseness of voice and swelling of tongue. Most likely diagnosis is:
Which of the following cardiac complications may develop in a 33 year old woman with systemic lupus erythematosus (SLE) because of her underlying condition?
All are true regarding Ankylosing spondylitis except -
TRUE about arthropathy in hemochromatosis:
Which of the following is true about psoriatic arthritis
Bechterew disease is?
Behcet's syndrome is characterized by all, Except:
Practice by Chapter
Rheumatoid Arthritis
Practice Questions
Spondyloarthropathies
Practice Questions
Systemic Lupus Erythematosus
Practice Questions
Vasculitis Syndromes
Practice Questions
Scleroderma and Related Disorders
Practice Questions
Inflammatory Myopathies
Practice Questions
Crystal Arthropathies
Practice Questions
Osteoarthritis
Practice Questions
Primary Immunodeficiency Disorders
Practice Questions
Autoinflammatory Syndromes
Practice Questions
Sjögren's Syndrome
Practice Questions
Antiphospholipid Syndrome
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app