
Rheumatology and Immunology — MCQs

On this page
A 35-year-old female presents with skin thickening and muscle weakness. Her peripheries became pale on exposure to cold. Her ANA is positive and creatine kinase is increased. Scl-70 is positive and perifascicular infiltration is noted in biopsy. What is the antibody associated with this condition?
History of a woman with skin features like limited cutaneous systemic sclerosis (scleroderma) was given. What is the most specific marker?
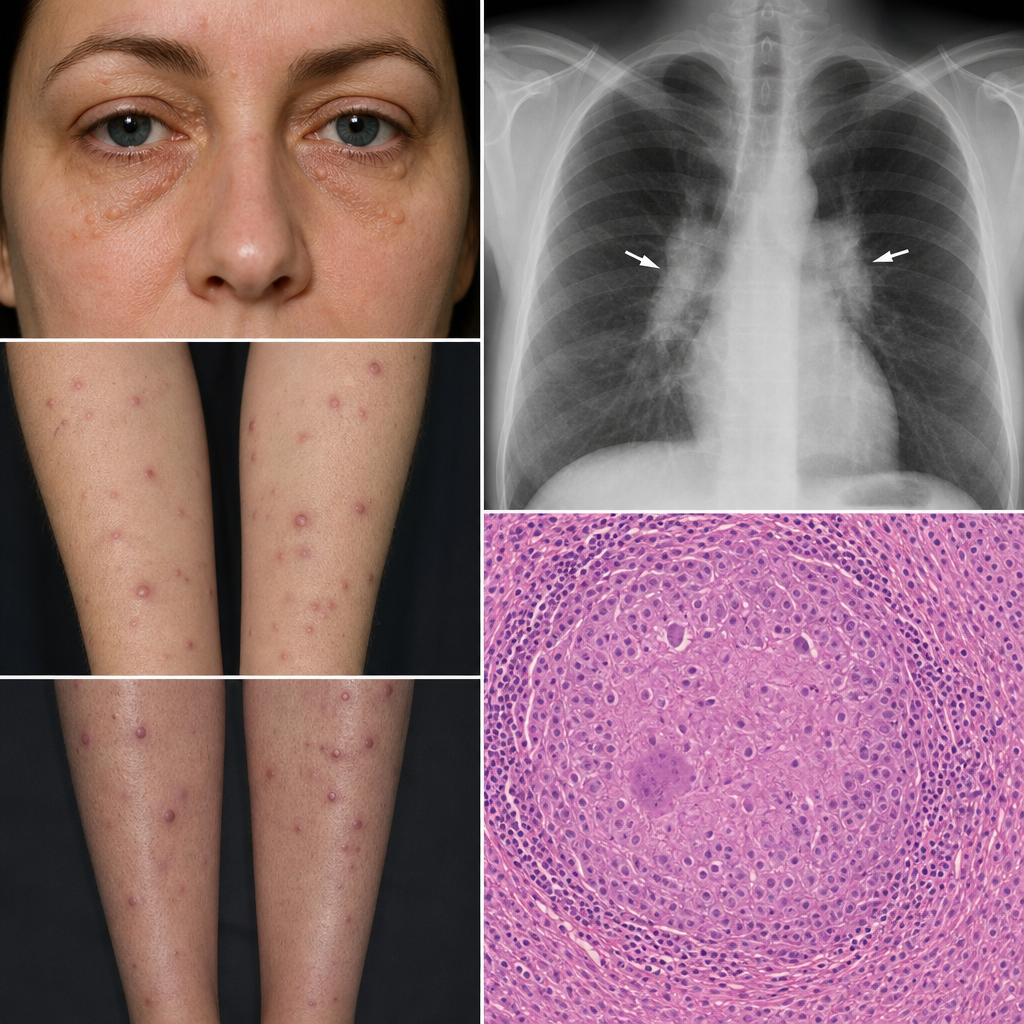
A 35-year-old woman presents with multiple painless nodules on her shins, forearms, and around her eyes. Biopsy shows non-caseating granulomas. Chest X-ray reveals bilateral hilar lymphadenopathy. Which of the following is most likely to be elevated in this condition?
A 48-year-old female presents to your office with a 1-year history of dry eyes and difficulty swallowing. She complains of blinking frequently and of eye strain while using her computer at work. She also reports stiffness in her knees and lower back. Past medical history is unremarkable and she does not take medications. She denies cigarette or alcohol use. Family history is notable for Hashimoto's thyroiditis in her mother. Physical exam shows dry oral mucosa and enlargement of the parotid glands. Which of the following serologies is likely to be positive in this patient?
A lady, when exposed to cold, experiences extremities turning blue. Which of the following antibodies is associated with this condition?
Which of the following is the best investigation for acute gout?
Diagnosis of Gout is confirmed by which test?
Primary osteoarthritis affects all except:
A 52-year-old woman has long-standing rheumatoid arthritis (RA) and is being treated with corticosteroids and nonsteroidal anti-inflammatory drugs (NSAIDs). Which of the following cardiac complications may arise in this clinical setting?
Arthritis mutilans is seen in?
Practice by Chapter
Rheumatoid Arthritis
Practice Questions
Spondyloarthropathies
Practice Questions
Systemic Lupus Erythematosus
Practice Questions
Vasculitis Syndromes
Practice Questions
Scleroderma and Related Disorders
Practice Questions
Inflammatory Myopathies
Practice Questions
Crystal Arthropathies
Practice Questions
Osteoarthritis
Practice Questions
Primary Immunodeficiency Disorders
Practice Questions
Autoinflammatory Syndromes
Practice Questions
Sjögren's Syndrome
Practice Questions
Antiphospholipid Syndrome
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app