
Nephrology — MCQs

On this page
A 40-year-old male with known cirrhosis has serum bilirubin 2.5 mg/dL, albumin 3.0 g/dL, PT prolonged by 4 seconds, grade I ascites, and no encephalopathy. What is his Child-Pugh score class?
Antibody-coated bacteria are characteristic of which one of the following conditions?
A 36-year-old male presents with a complaint of passing dark-reddish urine. He states that yesterday he played basketball for 4 hours, which was the first time he had exercised in 4 months. He awoke this morning with sore muscles and discolored urine. Physical examination is unremarkable. The urine is reddish-brown in color; dipstick test for blood is positive, the pH is 5.1, the specific gravity 1.03. Microscopic examination of the urine reveals no red blood cells. What is the most likely etiology for this presentation?
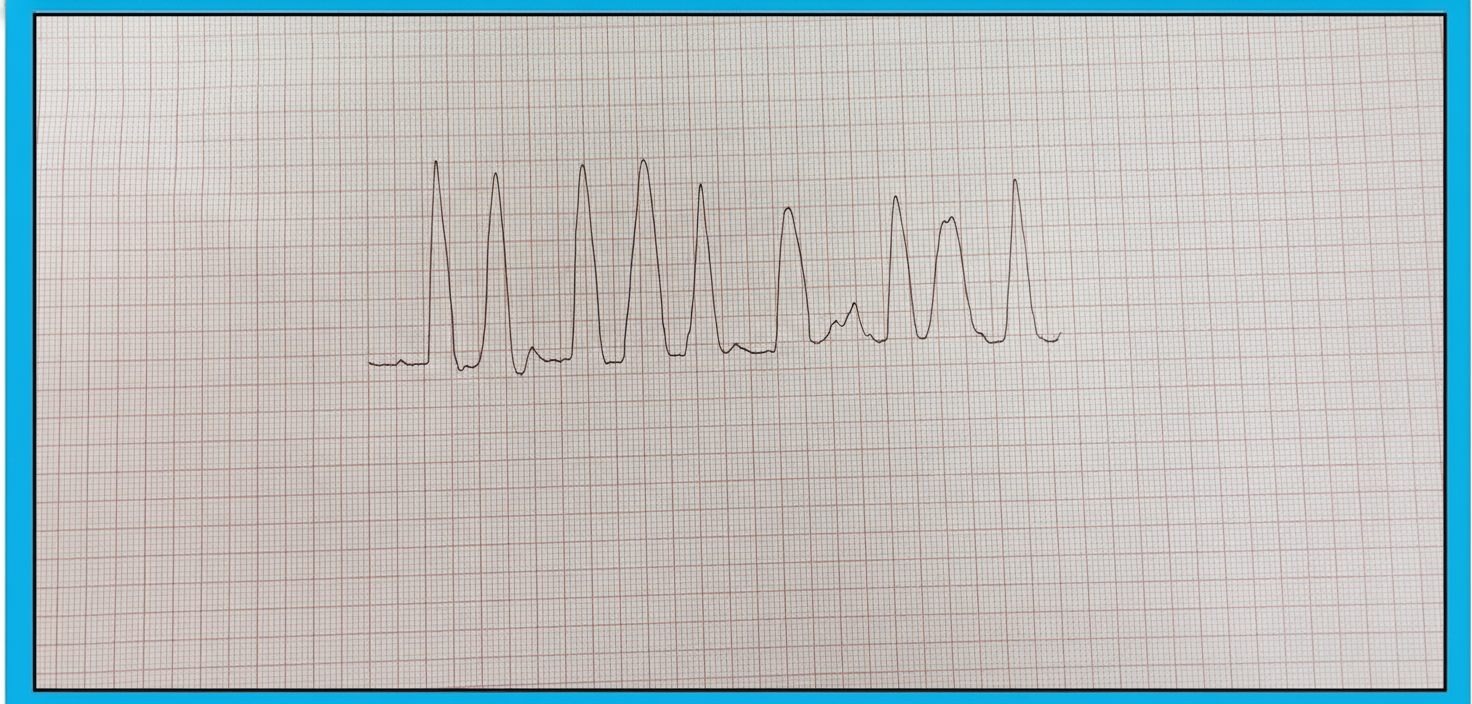
A 78-year-old man with advanced renal disease has the ECG (lead II). What is the diagnosis?
A patient presents to a clinic with complaints of headache and fatigue. Lab data show serum sodium, 122 mEq/L; serum osmolality, 240 mOsm/L; urine osmolality, 455 mOsm/L. Which condition best correlates with these data?
Practice by Chapter
Acute Kidney Injury
Practice Questions
Chronic Kidney Disease
Practice Questions
Glomerular Diseases
Practice Questions
Tubulointerstitial Diseases
Practice Questions
Nephrotic and Nephritic Syndromes
Practice Questions
Urinary Tract Infections
Practice Questions
Renal Replacement Therapy
Practice Questions
Fluid and Electrolyte Disorders
Practice Questions
Acid-Base Disorders
Practice Questions
Kidney in Systemic Diseases
Practice Questions
Kidney Stones and Obstructive Uropathy
Practice Questions
Hypertension in Kidney Disease
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app