
Hematology — MCQs

On this page
Gum hypertrophy is a clinical feature of which of the following conditions?
Which of the following statements regarding hereditary hemorrhagic telangiectasia is true?
Christmas disease is treated by?
A 30-year-old female presents with an RBC count of 4.5 million/µL, MCV of 55 fL, and TLC of 8000/µL. She has no history of blood transfusion. What is the most likely diagnosis?
A 30-year-old male presented with a skin lesion in his right axilla. A presumptive diagnosis of staphylococcal skin carbuncle was made and the patient was treated with empiric trimethoprim/sulfamethoxazole. After 2 days, the patient presented to the emergency department with excessive weakness, abdominal pain, and dark-colored urine. On examination, vital signs were normal, and jaundice was present. Laboratory studies showed a drop in Hb from 14 g/dl to 8 g/dl and a rise in bilirubin levels. Urine dipstick was positive for bilirubin. Peripheral blood smear findings are suggestive of a specific condition. What is the most likely diagnosis for this patient?
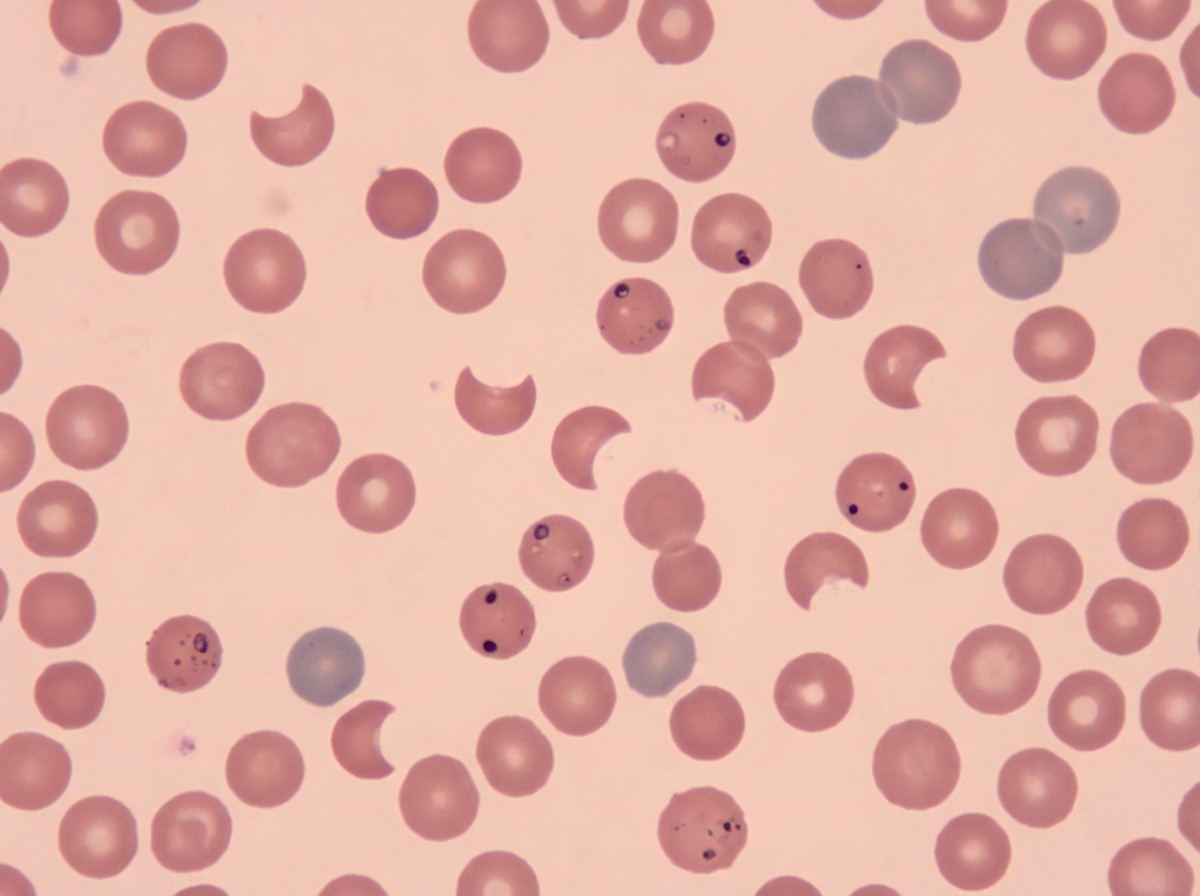
A patient presents with macroglossia and decreased hemoglobin. Laboratory investigations reveal low vitamin B12 levels and low folic acid levels. What is the most likely diagnosis?
What is the most appropriate drug used in the management of iron chelation in beta-thalassemia major?
A crisis in a patient with sickle cell disease is most likely to be caused by which of the following conditions?
Classical "Rain drop" lesions are seen in which of the following conditions?
Which of the following is true regarding lymph node enlargement in Hodgkin's Lymphoma?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app