
Hematology — MCQs

On this page
Which of the following is not included in the Gaisböck syndrome?
A 38-year-old female presents with fatigue, weakness, and pallor for 3 months. She has a history of menorrhagia. Laboratory findings show hemoglobin 8.5 g/dL, MCV 68 fL, MCH 22 pg, serum ferritin 8 ng/mL, serum iron 30 μg/dL, TIBC 450 μg/dL, and peripheral smear shows microcytic hypochromic RBCs with pencil cells. Bone marrow examination shows absent iron stores. What is the most appropriate next step in management?
A 32-year-old woman presents with easy bruising and petechiae. Her platelet count is 18,000 / mm³. Hemoglobin and total leukocyte count are normal. She recently recovered from a mild upper respiratory tract infection. Peripheral smear shows no abnormal cells. What is the most appropriate next step in management?
A 16-year-old boy presents with jaundice and splenomegaly. His father had a similar illness during adolescence. MCHC : high? What is the most likely diagnosis?
Which of the following is the most common inherited bleeding disorder?
The following test is performed for which condition?

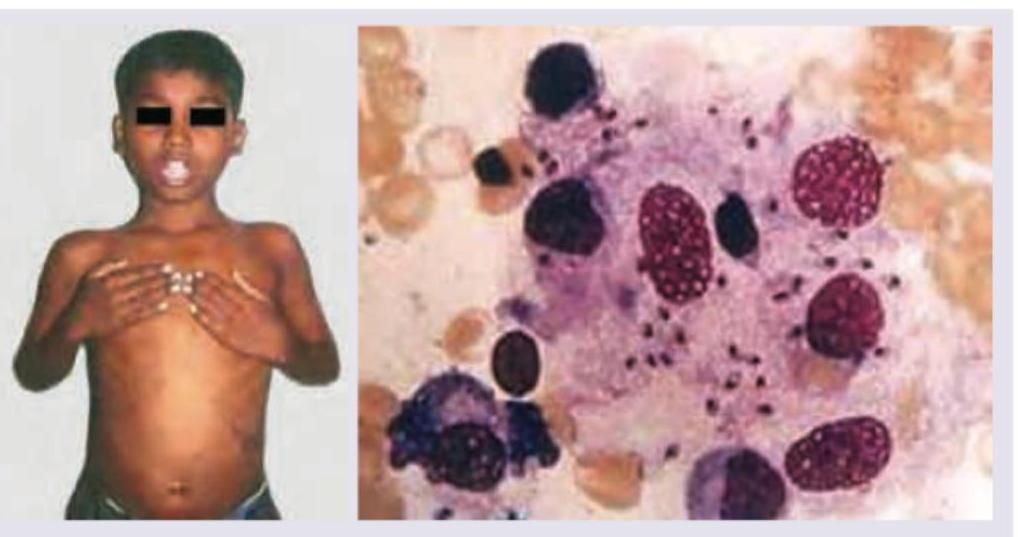
An 8-year-old child of migrant construction worker presents with fever for last 2 weeks with progressive pallor. Complete blood counts reveal pancytopenia, malaria work up is negative, Total serum protein is 7 gm% with Serum Globulin of 4.5 gm%, serum albumin 3.5 mg%. Bone marrow study shows?
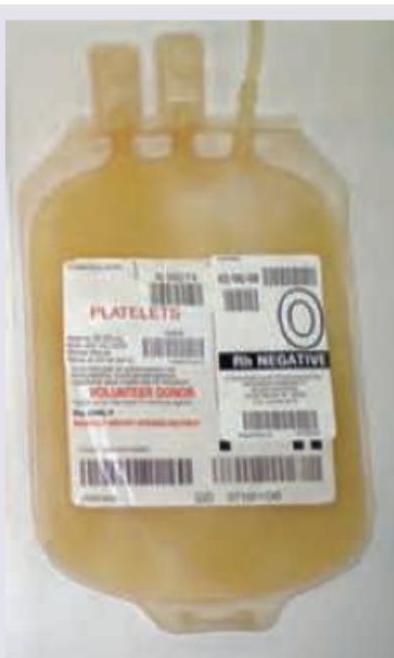
All are true about the blood component shown below except?
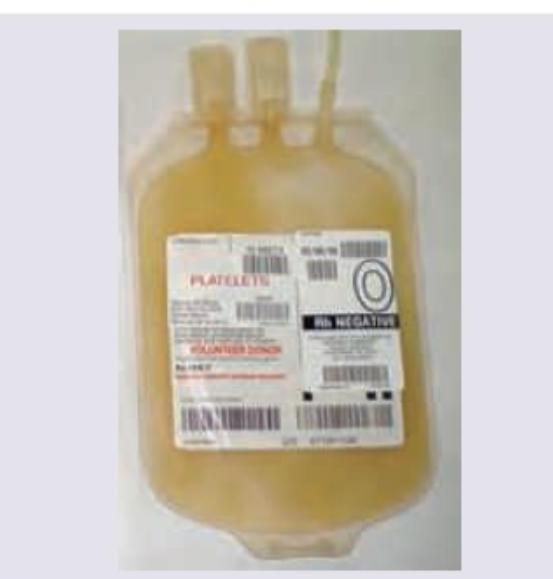
What is the threshold for prophylactic administration of the following blood product?
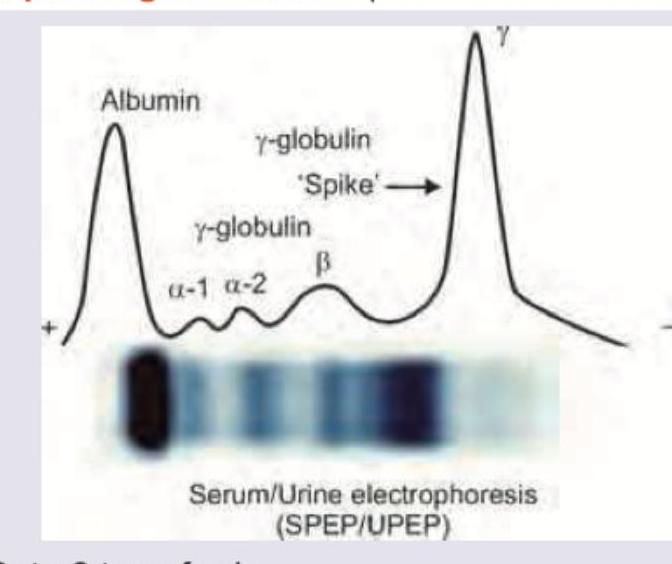
Which of the following is a prognostic marker for the patient whose urinary and serum electrophoresis report is given below?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app