
Hematology — MCQs

On this page
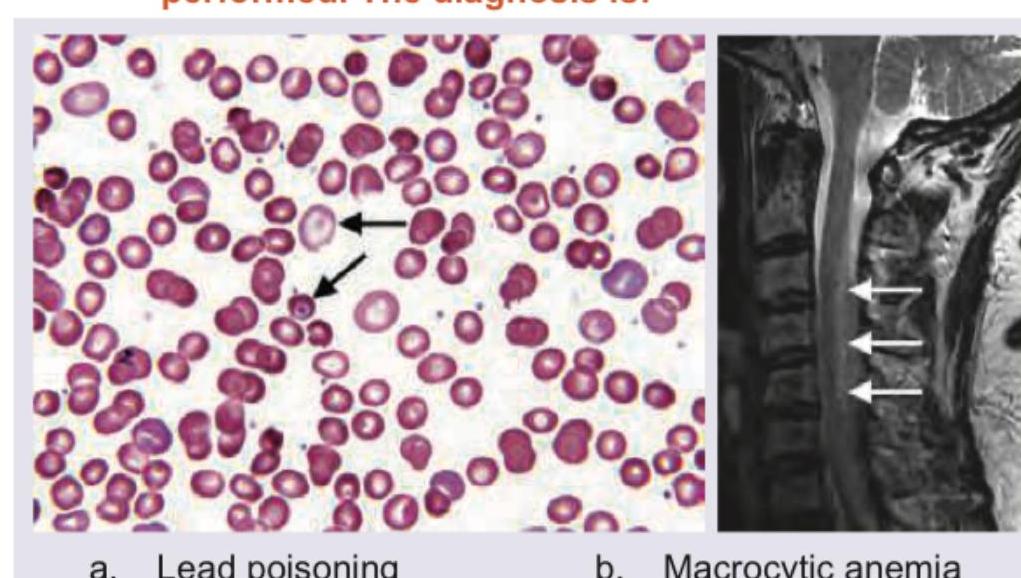
A 45-year-old person has complaints of paresthesia in hand and feet with progressive spastic weakness. On examination, absent ankle jerk with Babinski sign is noted. Peripheral smear and MRI spine were performed. The diagnosis is:
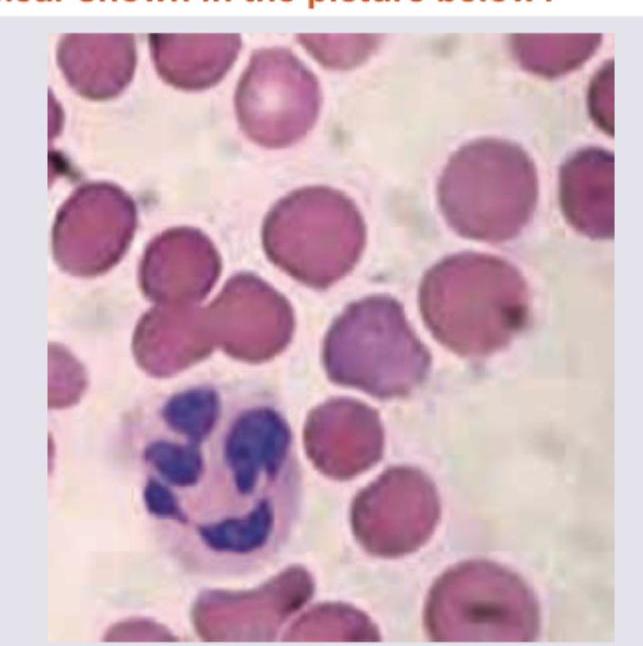
Which is shown in the peripheral smear of patient with metallic heart prosthesis?
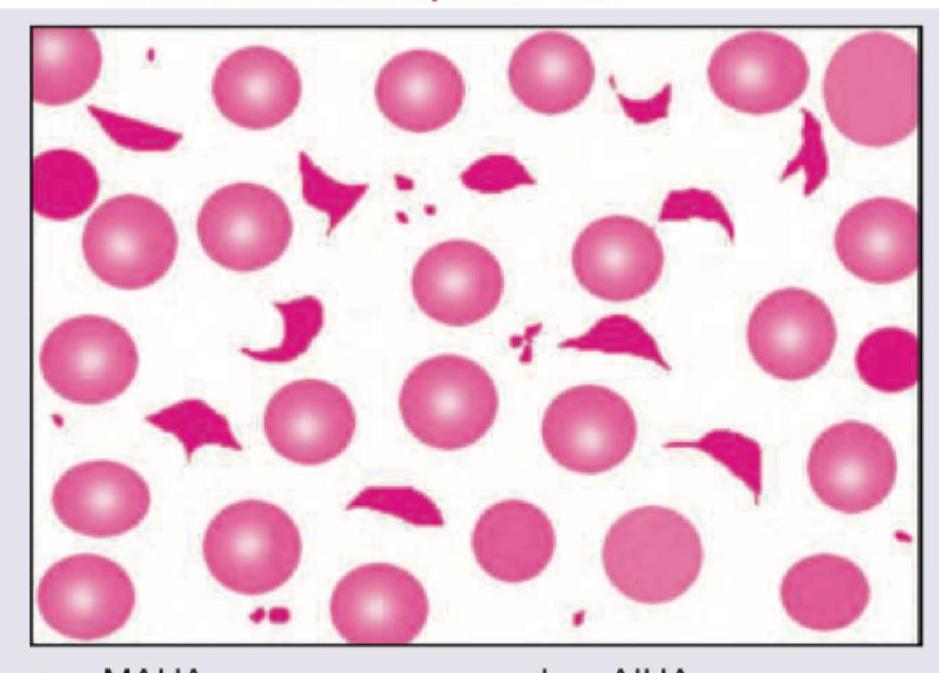
Which of the following is seen with the peripheral smear shown in the picture below?
Which of the following treatments are recommended for a pregnant woman suffering from sickle cell disease ? I. Folic acid 1 mg daily II. Azathioprine III. Penicillin prophylaxis IV. Thromboprophylaxis with low molecular weight heparin Select the correct answer using the code given below :
Which of the following are secondary iron overload conditions? I. Transfusion related iron load II. Thalassemia III. Hepatitis C associated liver disease Select the correct answer using the code given below :
With regard to management of idiopathic thrombocytopenic purpura, consider the following statements : I. All patients with ITP with platelet count less than 100 x 10^9 / L should receive high dose of glucocorticoids II. For patient with spontaneous bleeding, 40 mg of dexamethasone daily is indicated for 4 days III. Intravenous immunoglobulin can raise the platelet count IV. Life threatening bleeding should be treated with platelet transfusion Which of the statements given above are correct?
First line therapy in chronic phase of chronic myeloid leukemia is
Which one of the following causes low-volume erythrocytosis?
A 35 year female presents with easy fatigability. Investigations show that Hb is 6 g %; Red cell morphology is normocytic normochromic; and reticulocyte production index is 5.5 . Which one of the following conditions favours this abnormality?
Which of the following are blood values of Iron Deficiency Anaemia ? 1. Serum iron is less than 30 mg/100 mL 2. Total iron binding capacity is less than 400 µg/mL 3. Percentage saturation is 10% or less 4. Serum ferritin is below 30 µg/mL Select the correct answer using the code given below :
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app