
Hematology — MCQs

On this page
Iron overload occurs in all of the following conditions except?
Polycythemia is not caused by which of the following?
A 23-year-old asymptomatic female pilot presents with MCV of 70 fL, ferritin of 100 g/L, and Hb of 10 g/dL. What is the most likely cause?
Hemophilia manifests clinically as a rise in which of the following parameters?
A patient presented with painless proptosis. What is the next investigation to diagnose it as chloroma?
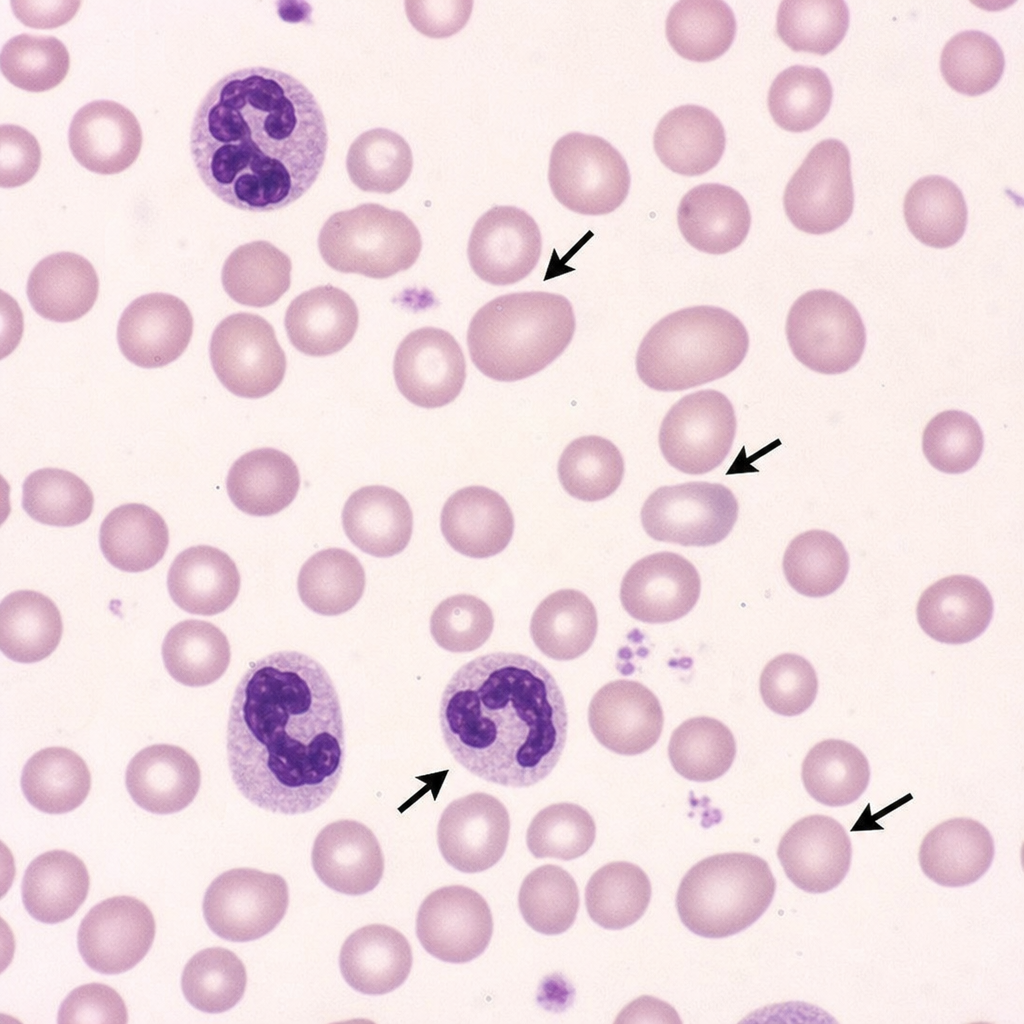
A 20-year-old patient presents with a year of fatigue and tiredness. Investigations show Hb 9g/dL, MCV 10%, and peripheral smear reveals macrocytic red blood cells with hypersegmented neutrophils. What is the most likely diagnosis?
Peripheral smear showing the arrows is diagnostic of which of the following conditions?
What is the most appropriate investigation for a 40-year-old female presenting with anemia, jaundice, and spherocytosis?
What is the most common inherited bleeding disorder?
Which one of the following findings is NOT consistent with the diagnosis of aplastic anemia?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app