
Hematology — MCQs

On this page
A 23-year-old female presented with jaundice and pallor for 2 months. Her peripheral blood smear shows the presence of spherocytes. What is the most relevant investigation to arrive at a diagnosis?
What is the most common site of thrombosis in paroxysmal nocturnal hemoglobinuria?
A 67-year-old man presents with 5 months of increasing weakness, fatigue, and weight loss. He has experienced decreasing vision in both eyes, headaches, and dizziness. His hands are sensitive to cold. Physical examination reveals generalized lymphadenopathy and hepatosplenomegaly. Laboratory studies show a serum protein level of 15.5 g/dL and an albumin concentration of 3.2 g/dL. A bone marrow biopsy shows infiltration by numerous small plasmacytoid lymphoid cells with Russell bodies in the cytoplasm. Which of the following additional laboratory findings is most likely to be reported for this patient?
Which of the following factors can differentiate primary polycythemia from secondary polycythemia?
What is the most likely subtype of leukemia associated with the provided finding?
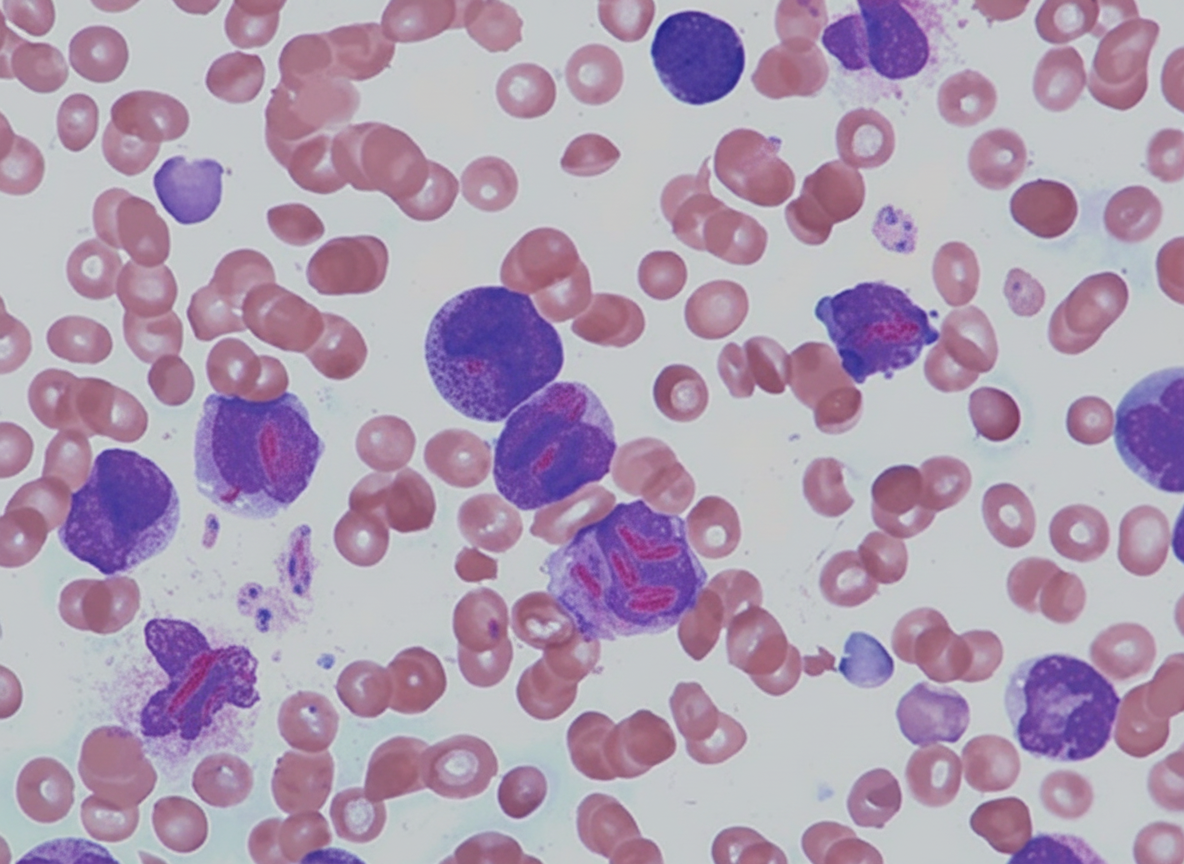
Plasmapheresis is recommended in all of the following conditions except?
A 60-year-old man presents with a 6-month history of increasing fatigue. Physical examination reveals marked pallor, and a CBC shows a macrocytic anemia. Which of the following is the most likely cause of anemia in this patient?
What is the commonest acute presentation of sickle cell anemia?
Which of the following conditions has the least impact on life expectancy since the point of diagnosis?
A 48-year-old woman presents with a 3-week history of fatigue, yellow skin, and yellow sclerae. Physical examination reveals mild jaundice. Her serum bilirubin level is 3.7 mg/dL, predominantly unconjugated. Liver function tests, including serum AST, ALT, and alkaline phosphatase, are normal. Hemoglobin level is 6.0 g/dL. Jaundice resolves after corticosteroid administration. What is the most likely cause of hyperbilirubinemia in this patient?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app