
Hematology — MCQs

On this page
Which of the following is true about aplastic anemia?
Pancytopenia with cellular marrow is seen in which of the following conditions?
A 38-year-old woman presents with repeated episodes of sore throat. She is on no medications, does not use ethanol, and has no history of renal disease. Physical examination is normal. Hemoglobin is 9.0 g/dL, mean corpuscular volume is 85 fL, white blood cell count is 2000/mL, and platelet count is 30,000/mL. Which of the following is the best approach to diagnosis?
Which tumor is associated with polycythemia?
A 16-year-old female presents with generalized weakness and palpitations. Her Hb is 7 g/dL and peripheral smear shows microcytic hypochromic anemia. Reticulocyte count is 0.8%. Serum bilirubin is 1 mg%. What is the most likely diagnosis?
A 69-year-old woman presents with a hip fracture sustained from a fall. She has a history of recurrent pneumonia over the past year. Laboratory findings include a normal white blood cell count, thrombocytopenia, and an elevated erythrocyte sedimentation rate (ESR). X-rays show multiple lytic bone lesions. Serum electrophoresis reveals an M-protein spike. What is the most likely diagnosis?
Hepatitis-associated aplastic anemia is caused by which type of hepatitis virus?
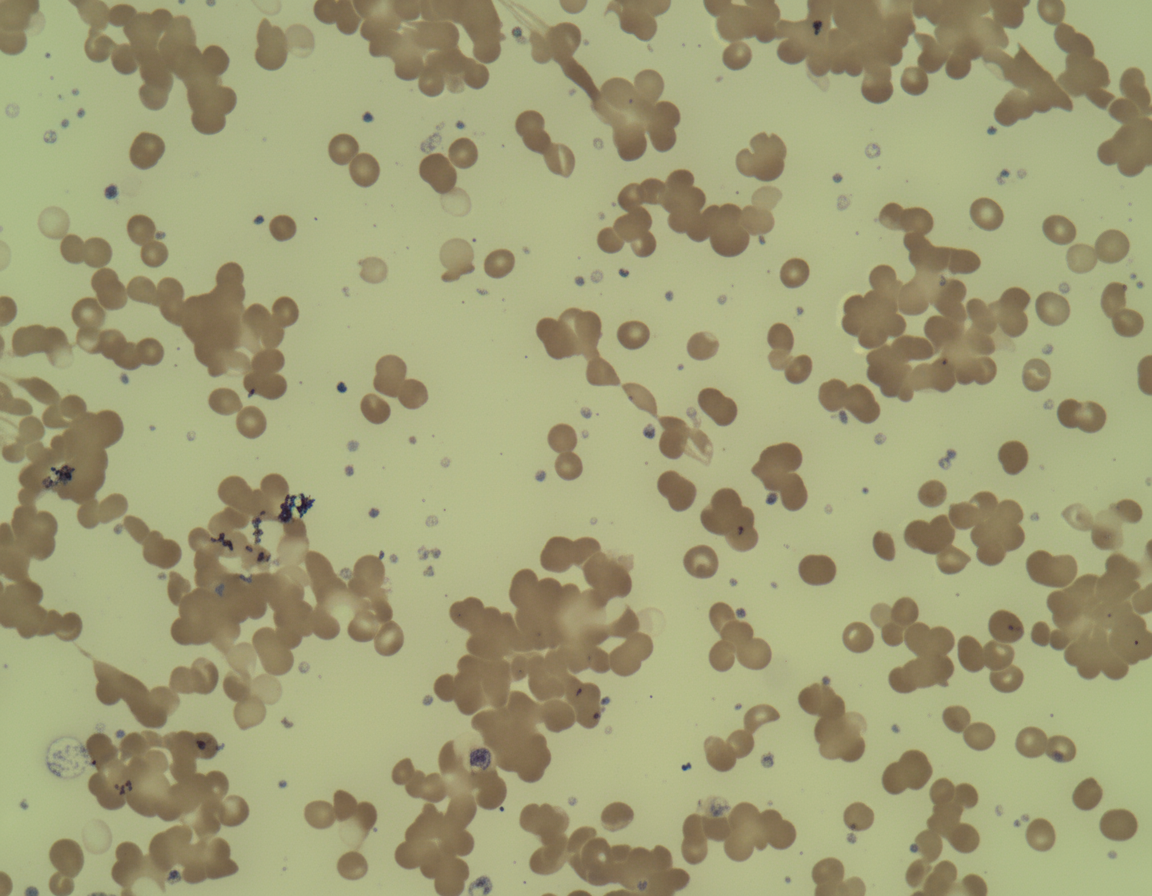
A 25-year-old female presented in December with chronic fatigue and cyanosis with bluish lips and arthralgia. Peripheral blood film is shown below. What is the likely cause?
A patient presents with increased serum ferritin, decreased TIBC, increased serum iron, and increased % saturation. What is the most probable diagnosis?
A young person presents with a history of severe menorrhagia, palpable spleen, prolonged bleeding time, and normal clotting time and platelet count. What is the most likely diagnosis?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app