
Hematology — MCQs

On this page
Which of the following does not cause megaloblastic anemia?
A 43-year-old woman complains of constant tiredness, light-headedness, and occasional palpitations and shortness of breath. Physical examination shows pallor of the oral mucosa and glossitis. Neurologic examination reveals paresthesias, numbness, decreased vibration sensation, and loss of deep tendon reflexes. Laboratory studies include hemoglobin of 7.2 g/dL, WBC of 4,500/mL, platelets of 140,000/mL, erythrocyte folate of 220 ng/mL, serum vitamin B12 of 40 pg/mL, serum anti-intrinsic factor of 1:128, and serum anti-parietal cell antibody of 1:64. Peripheral blood shows macrocytic anemia with poikilocytosis of RBCs and hypersegmented neutrophils. Atrophic gastritis is diagnosed by gastric biopsy. What is the expected bone marrow examination finding in this patient?
All of the following are true about Chronic Lymphocytic Leukemia, except?
A 30-year-old man presents with anemia, clinical jaundice, and splenomegaly. He has an autosomal dominant disorder for which splenectomy is the only treatment. What is the most likely diagnosis?
What is the primary abnormality in the sequence of events leading to Disseminated Intravascular Coagulation (DIC)?
All are true about thrombotic thrombocytopenic purpura except?
A 63-year-old woman experiences a burning sensation in her hands and feet. Two months prior, she had an episode of swelling with tenderness in the right leg, followed by dyspnea and right-sided chest pain. Physical examination reveals splenomegaly and hepatomegaly. CBC shows hemoglobin, 13.3 g/dL; hematocrit, 40.1%; MCV, 91 mm3; platelet count, 657,000/mm3; and WBC count, 17,400/mm3. The peripheral blood smear shows abnormally large platelets. Which of the following is the most likely diagnosis?
A patient presents with a hemoglobin of 5 g%, total leukocyte count of 9000/cc, differential leukocyte count showing 3% neutrophils and 75% lymphoblasts, and has had a fever for 1 month. Which of the following management options would be appropriate?
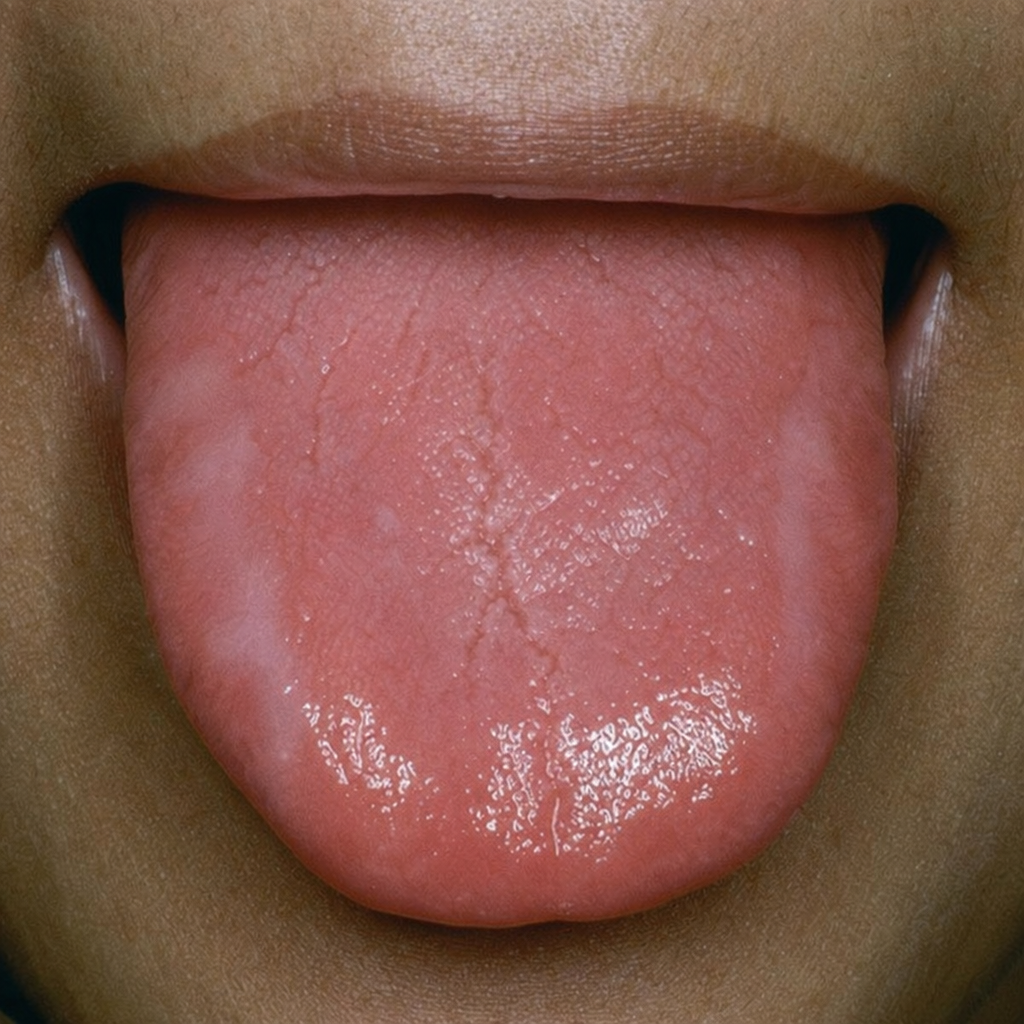
In which condition is the following finding in the tongue commonly seen?
Which of the following parameters will fall the earliest in iron deficiency?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app